

Autosomal dominant eccentric core disease caused by a heterozygous mutation in the MYH7 gene
- PMID: 24828896
- PMCID: PMC4173876
- DOI: 10.1136/jnnp-2013-306754
Autosomal dominant eccentric core disease caused by a heterozygous mutation in the MYH7 gene
Abstract
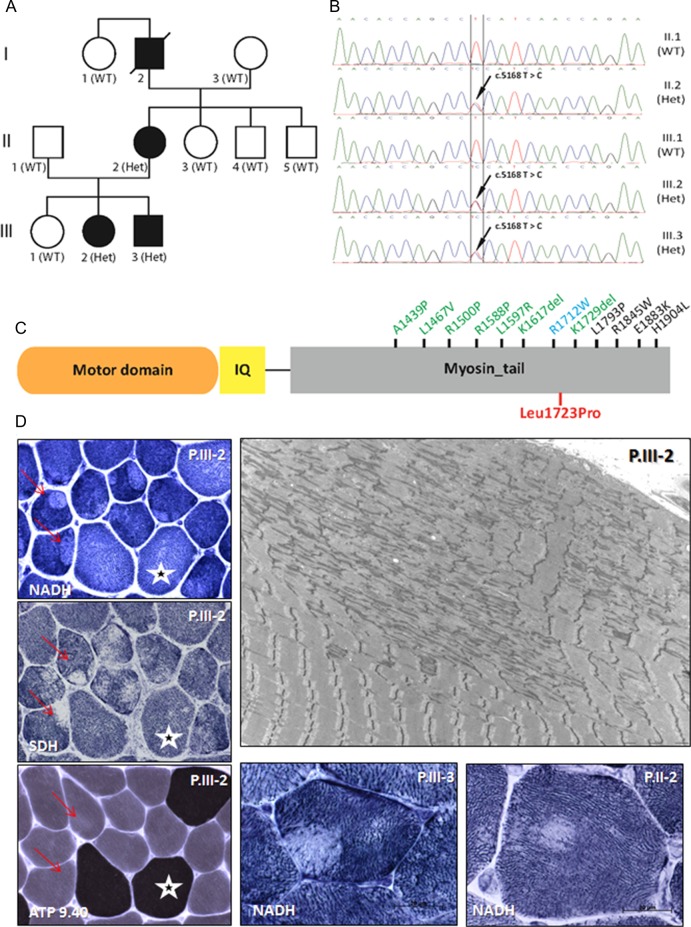
Background: Autosomal dominant (AD) central core disease (CCD) is a congenital myopathy characterised by the presence of cores in the muscle fibres which correspond to broad areas of myofibrils disorganisation, Z-line streaming and lack of mitochondria. Heterozygous mutations in the RYR1 gene were observed in the large majority of AD-CCD families; however, this gene was excluded in some of AD-CCD families.
Objective: To enlarge the genetic spectrum of AD-CCD demonstrating mutations in an additional gene.
Patients and methods: Four affected AD family members over three generations, three of whom were alive and participate in the study: the mother and two of three siblings. The symptoms began during the early childhood with mild delayed motor development. Later they developed mainly tibialis anterior weakness, hypertrophy of calves and significant weakness (amyotrophic) of quadriceps. No cardiac or ocular involvement was noted.
Results: The muscle biopsies sections showed a particular pattern: eccentric cores in type 1 fibres, associated with type 1 predominance. Most cores have abrupt borders. Electron microscopy confirmed the presence of both unstructured and structured cores. Exome sequencing analysis identified a novel heterozygous missense mutation p.Leu1723Pro in MYH7 segregating with the disease and affecting a conserved residue in the myosin tail domain.
Conclusions: We describe MYH7 as an additional causative gene for AD-CCD. These findings have important implications for diagnosis and future investigations of AD-congenital myopathies with cores, without cardiomyopathy, but presenting a particular involvement of distal and quadriceps muscles.
Keywords: MUSCLE DISEASE; MYOPATHY; NEUROPATHOLOGY, MUSCLE.
Published by the BMJ Publishing Group Limited. For permission to use (where not already granted under a licence) please go to http://group.bmj.com/group/rights-licensing/permissions.
Figures

Similar articles
-
Mutations in MYH7 cause Multi-minicore Disease (MmD) with variable cardiac involvement.Neuromuscul Disord. 2012 Dec;22(12):1096-104. doi: 10.1016/j.nmd.2012.06.007. Epub 2012 Jul 10. Neuromuscul Disord. 2012. PMID: 22784669
-
Novel slow-skeletal myosin (MYH7) mutation in the original myosin storage myopathy kindred.Neuromuscul Disord. 2006 Jun;16(6):357-60. doi: 10.1016/j.nmd.2006.03.011. Epub 2006 May 8. Neuromuscul Disord. 2006. PMID: 16684601
-
A novel heterozygous missense MYH7 mutation potentially causes an autosomal dominant form of myosin storage myopathy with dilated cardiomyopathy.BMC Cardiovasc Disord. 2023 Oct 4;23(1):487. doi: 10.1186/s12872-023-03538-8. BMC Cardiovasc Disord. 2023. PMID: 37794383 Free PMC article.
-
A recurrent single-amino acid deletion (p.Glu500del) in the head domain of ß-cardiac myosin in two unrelated boys presenting with polyhydramnios, congenital axial stiffness and skeletal myopathy.Orphanet J Rare Dis. 2022 Jul 19;17(1):279. doi: 10.1186/s13023-022-02421-7. Orphanet J Rare Dis. 2022. PMID: 35854315 Free PMC article. Review.
-
Hereditary myosin myopathies.Neuromuscul Disord. 2007 May;17(5):355-67. doi: 10.1016/j.nmd.2007.02.008. Epub 2007 Apr 16. Neuromuscul Disord. 2007. PMID: 17434305 Review.
Cited by
-
The sarcomeric M-region: a molecular command center for diverse cellular processes.Biomed Res Int. 2015;2015:714197. doi: 10.1155/2015/714197. Epub 2015 Apr 15. Biomed Res Int. 2015. PMID: 25961035 Free PMC article. Review.
-
Mutations in proteins involved in E-C coupling and SOCE and congenital myopathies.J Gen Physiol. 2022 Sep 5;154(9):e202213115. doi: 10.1085/jgp.202213115. Epub 2022 Aug 18. J Gen Physiol. 2022. PMID: 35980353 Free PMC article. Review.
-
Myoimaging in the NGS era: the discovery of a novel mutation in MYH7 in a family with distal myopathy and core-like features--a case report.BMC Med Genet. 2016 Mar 22;17:25. doi: 10.1186/s12881-016-0288-0. BMC Med Genet. 2016. PMID: 27005958 Free PMC article.
-
MYH7 in cardiomyopathy and skeletal muscle myopathy.Mol Cell Biochem. 2024 Feb;479(2):393-417. doi: 10.1007/s11010-023-04735-x. Epub 2023 Apr 20. Mol Cell Biochem. 2024. PMID: 37079208 Review.
-
'Dusty core disease' (DuCD): expanding morphological spectrum of RYR1 recessive myopathies.Acta Neuropathol Commun. 2019 Jan 5;7(1):3. doi: 10.1186/s40478-018-0655-5. Acta Neuropathol Commun. 2019. PMID: 30611313 Free PMC article.
References
-
- Romero NB, Herasse M, Monnier N, et al. Clinical and histopathological aspects of Central Core Disease associated and non-associated with RYR1 locus. Acta Myologica 2005;XXIV:70–3 - PubMed
-
- Fardeau M. Congenital myopathies. In: Mastaglia FL, Walton Sir John, eds. Skeletal Muscle pathology. Chap. 4 Edinburgh, London, Melbourne and New York: Churchill Livingstone, 1982
-
- Pegoraro E, Gavassini BF, Borsato C, et al. MYH7 gene mutation in myosin storage myopathy and scapulo-peroneal myopathy. Neuromusc Disord 2007;17:321–9 - PubMed
-
- Tasca G, Ricci E, Penttila S, et al. New phenotype and pathology features in MYH7-related distal myopathy. Neuromusc Disord 2012;22:640–7 - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources