

Targeting human apurinic/apyrimidinic endonuclease 1 (APE1) in phosphatase and tensin homolog (PTEN) deficient melanoma cells for personalized therapy
- PMID: 24830350
- PMCID: PMC4102809
- DOI: 10.18632/oncotarget.1926
Targeting human apurinic/apyrimidinic endonuclease 1 (APE1) in phosphatase and tensin homolog (PTEN) deficient melanoma cells for personalized therapy
Abstract
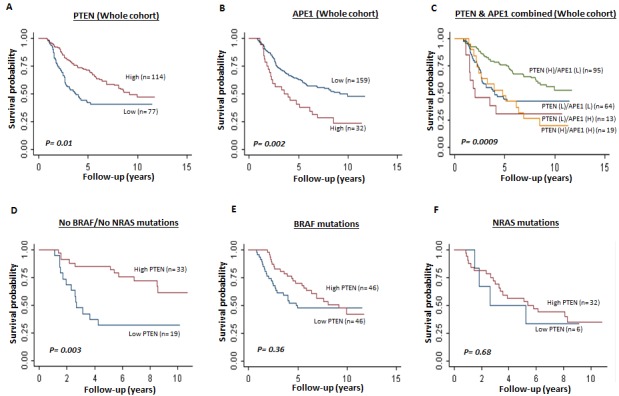
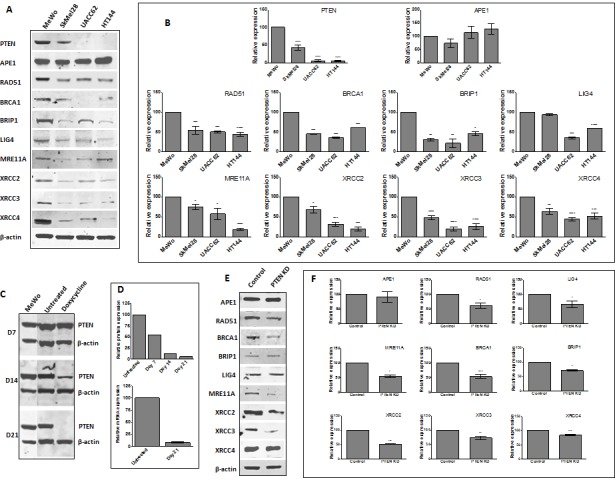
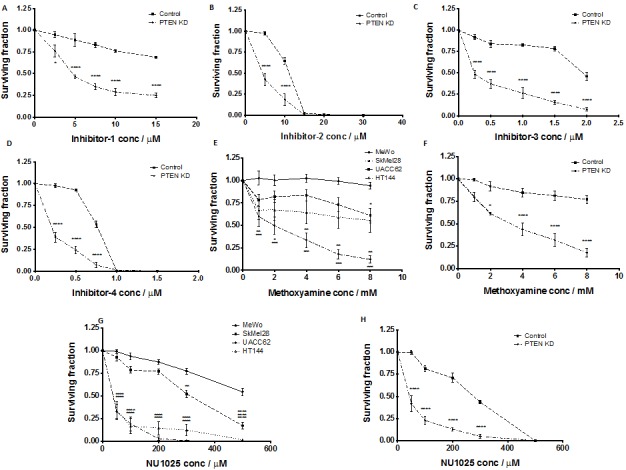
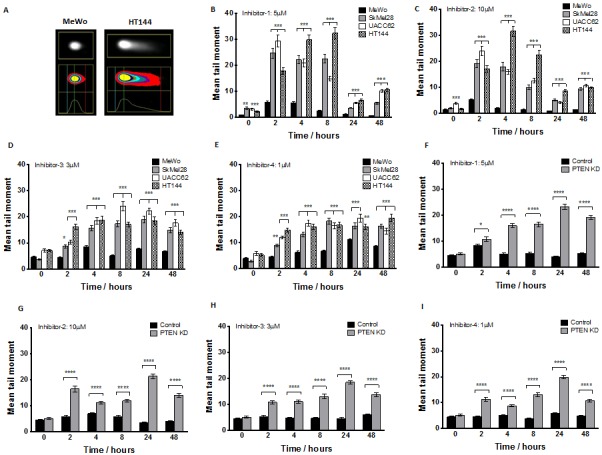
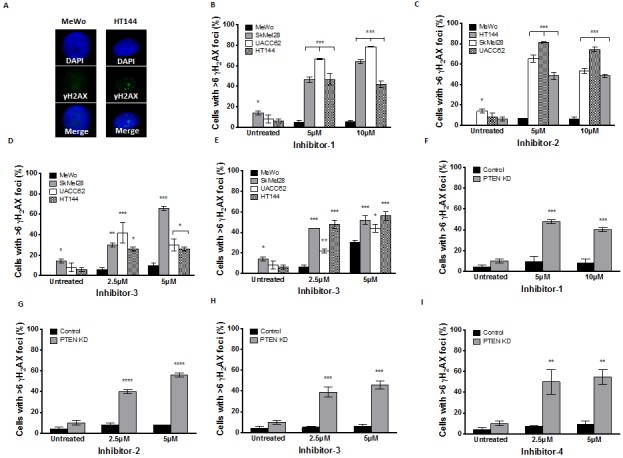
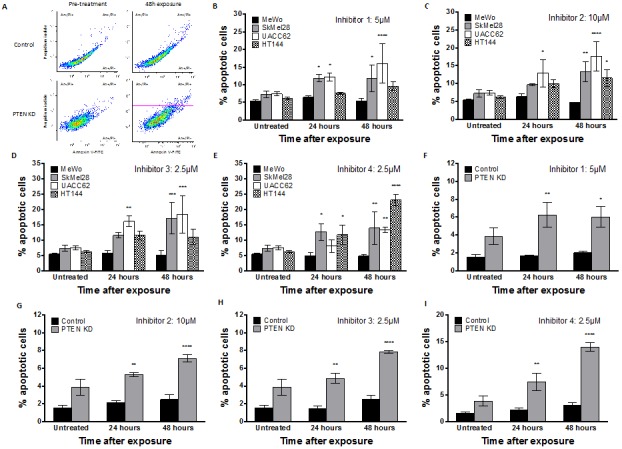
Phosphatase and tensin homolog (PTEN) loss is associated with genomic instability. APE1 is a key player in DNA base excision repair (BER) and an emerging drug target in cancer. We have developed small molecule inhibitors against APE1 repair nuclease activity. In the current study we explored a synthetic lethal relationship between PTEN and APE1 in melanoma. Clinicopathological significance of PTEN mRNA and APE1 mRNA expression was investigated in 191 human melanomas. Preclinically, PTEN-deficient BRAF-mutated (UACC62, HT144, and SKMel28), PTEN-proficient BRAF-wildtype (MeWo), and doxycycline-inducible PTEN-knockout BRAF-wildtype MeWo melanoma cells were DNA repair expression profiled and investigated for synthetic lethality using a panel of four prototypical APE1 inhibitors. In human tumours, low PTEN mRNA and high APE1 mRNA was significantly associated with reduced relapse free and overall survival. Pre-clinically, compared to PTEN-proficient cells, PTEN-deficient cells displayed impaired expression of genes involved in DNA double strand break (DSB) repair. Synthetic lethality in PTEN-deficient cells was evidenced by increased sensitivity, accumulation of DSBs and induction of apoptosis following treatment with APE1 inhibitors. We conclude that PTEN deficiency is not only a promising biomarker in melanoma, but can also be targeted by a synthetic lethality strategy using inhibitors of BER, such as those targeting APE1.
Conflict of interest statement
The authors disclose no potential conflicts of interest
Figures

References
-
- Abbotts R, Madhusudan S. Human AP endonuclease 1 (APE1): from mechanistic insights to druggable target in cancer. Cancer Treat Rev. 2010;36(5):425–435. - PubMed
-
- Dianov GL, Sleeth KM, Dianova, Allinson SL. Repair of abasic sites in DNA. Mutat Res. 2003;531(1-2):157–163. - PubMed
-
- Evans AR, Limp-Foster M, Kelley MR. Going APE over ref-1. Mutat Res. 2000;461(2):83–108. - PubMed
-
- Wang D, Xiang DB, Yang XQ, Chen LS, Li MX, Zhong ZY, Zhang YS. APE1 overexpression is associated with cisplatin resistance in non-small cell lung cancer and targeted inhibition of APE1 enhances the activity of cisplatin in A549 cells. Lung Cancer. 2009;66(3):298–304. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- R01 CA083115/CA/NCI NIH HHS/United States
- C8216/A6129/CRUK_/Cancer Research UK/United Kingdom
- R01 CA83115/CA/NCI NIH HHS/United States
- C588/A10589/CRUK_/Cancer Research UK/United Kingdom
- 1 R03 MH086444-01/MH/NIMH NIH HHS/United States
- G1000252/MRC_/Medical Research Council/United Kingdom
- C8216/A8168/CRUK_/Cancer Research UK/United Kingdom
- G0802123/MRC_/Medical Research Council/United Kingdom
- 11963/CRUK_/Cancer Research UK/United Kingdom
- C588/A4994/CRUK_/Cancer Research UK/United Kingdom
- 10589/CRUK_/Cancer Research UK/United Kingdom
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous
