

Bayesian test for colocalisation between pairs of genetic association studies using summary statistics
- PMID: 24830394
- PMCID: PMC4022491
- DOI: 10.1371/journal.pgen.1004383
Bayesian test for colocalisation between pairs of genetic association studies using summary statistics
Abstract
Genetic association studies, in particular the genome-wide association study (GWAS) design, have provided a wealth of novel insights into the aetiology of a wide range of human diseases and traits, in particular cardiovascular diseases and lipid biomarkers. The next challenge consists of understanding the molecular basis of these associations. The integration of multiple association datasets, including gene expression datasets, can contribute to this goal. We have developed a novel statistical methodology to assess whether two association signals are consistent with a shared causal variant. An application is the integration of disease scans with expression quantitative trait locus (eQTL) studies, but any pair of GWAS datasets can be integrated in this framework. We demonstrate the value of the approach by re-analysing a gene expression dataset in 966 liver samples with a published meta-analysis of lipid traits including >100,000 individuals of European ancestry. Combining all lipid biomarkers, our re-analysis supported 26 out of 38 reported colocalisation results with eQTLs and identified 14 new colocalisation results, hence highlighting the value of a formal statistical test. In three cases of reported eQTL-lipid pairs (SYPL2, IFT172, TBKBP1) for which our analysis suggests that the eQTL pattern is not consistent with the lipid association, we identify alternative colocalisation results with SORT1, GCKR, and KPNB1, indicating that these genes are more likely to be causal in these genomic intervals. A key feature of the method is the ability to derive the output statistics from single SNP summary statistics, hence making it possible to perform systematic meta-analysis type comparisons across multiple GWAS datasets (implemented online at http://coloc.cs.ucl.ac.uk/coloc/). Our methodology provides information about candidate causal genes in associated intervals and has direct implications for the understanding of complex diseases as well as the design of drugs to target disease pathways.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures

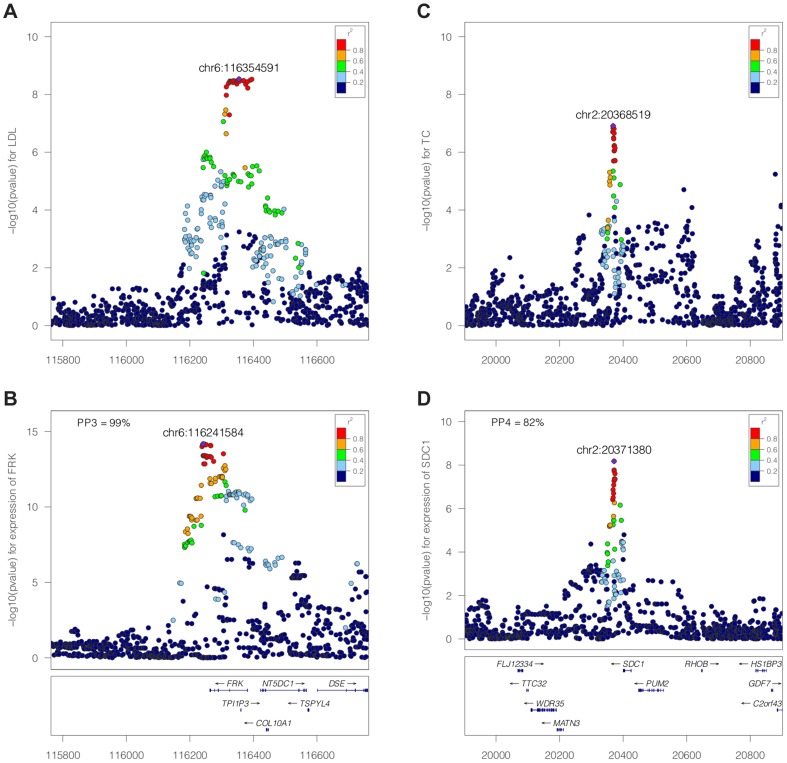
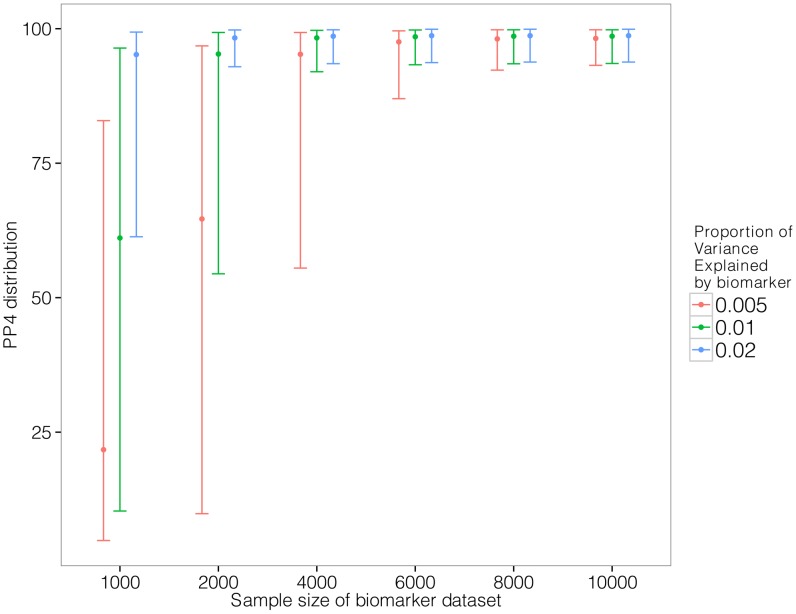
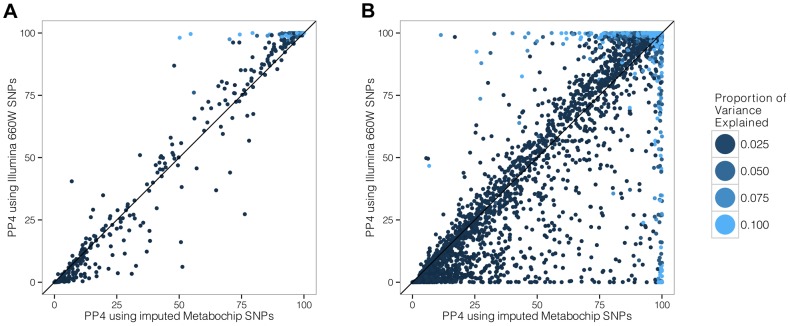
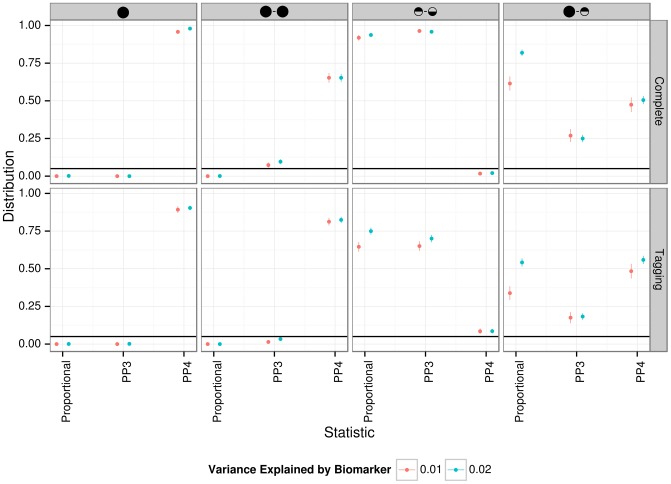

References
-
- Feero WG, Guttmacher AE, Manolio TA (2010) Genomewide association studies and assessment of the risk of disease. New England Journal of Medicine 363: 166–176. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
