

Evolving concepts in the pathogenesis of NASH: beyond steatosis and inflammation
- PMID: 24830559
- PMCID: PMC4057750
- DOI: 10.3390/ijms15058591
Evolving concepts in the pathogenesis of NASH: beyond steatosis and inflammation
Abstract
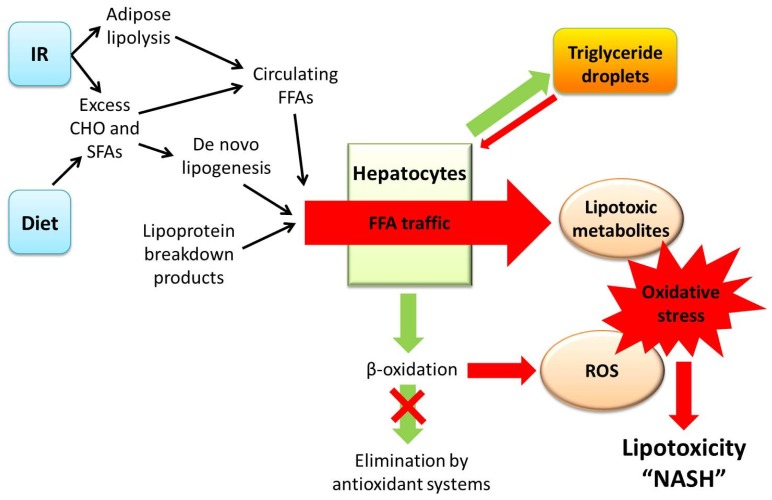
Non-alcoholic steatohepatitis (NASH) is characterised by hepatic steatosis and inflammation and, in some patients, progressive fibrosis leading to cirrhosis. An understanding of the pathogenesis of NASH is still evolving but current evidence suggests multiple metabolic factors critically disrupt homeostasis and induce an inflammatory cascade and ensuing fibrosis. The mechanisms underlying these changes and the complex inter-cellular interactions that mediate fibrogenesis are yet to be fully elucidated. Lipotoxicity, in the setting of excess free fatty acids, obesity, and insulin resistance, appears to be the central driver of cellular injury via oxidative stress. Hepatocyte apoptosis and/or senescence contribute to activation of the inflammasome via a variety of intra- and inter-cellular signalling mechanisms leading to fibrosis. Current evidence suggests that periportal components, including the ductular reaction and expansion of the hepatic progenitor cell compartment, may be involved and that the Th17 response may mediate disease progression. This review aims to provide an overview of the pathogenesis of NASH and summarises the evidence pertaining to key mechanisms implicated in the transition from steatosis and inflammation to fibrosis. Currently there are limited treatments for NASH although an increasing understanding of its pathogenesis will likely improve the development and use of interventions in the future.
Figures

References
-
- Clark J.M. The epidemiology of nonalcoholic fatty liver disease in adults. J. Clin. Gastroenterol. 2006;40:S5–S10. - PubMed
-
- Day C.P., James O.F. Steatohepatitis: A tale of two “hits”? Gastroenterology. 1998;114:842–845. - PubMed
-
- Tilg H., Moschen A.R. Evolution of inflammation in nonalcoholic fatty liver disease: The multiple parallel hits hypothesis. Hepatology. 2010;52:1836–1846. - PubMed
-
- Cortez-Pinto H., Chatham J., Chacko V.P., Arnold C., Rashid A., Diehl A.M. Alterations in liver ATP homeostasis in human nonalcoholic steatohepatitis: A pilot study. JAMA. 1999;282:1659–1664. - PubMed
-
- Mari M., Caballero F., Colell A., Morales A., Caballeria J., Fernandez A., Enrich C., Fernandez-Checa J.C., Garcia-Ruiz C. Mitochondrial free cholesterol loading sensitizes to TNF- and Fas-mediated steatohepatitis. Cell MeTable. 2006;4:185–198. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
