

Tyrosine 308 is necessary for ligand-directed Gs protein-biased signaling of β2-adrenoceptor
- PMID: 24831005
- PMCID: PMC4094047
- DOI: 10.1074/jbc.M114.558882
Tyrosine 308 is necessary for ligand-directed Gs protein-biased signaling of β2-adrenoceptor
Abstract
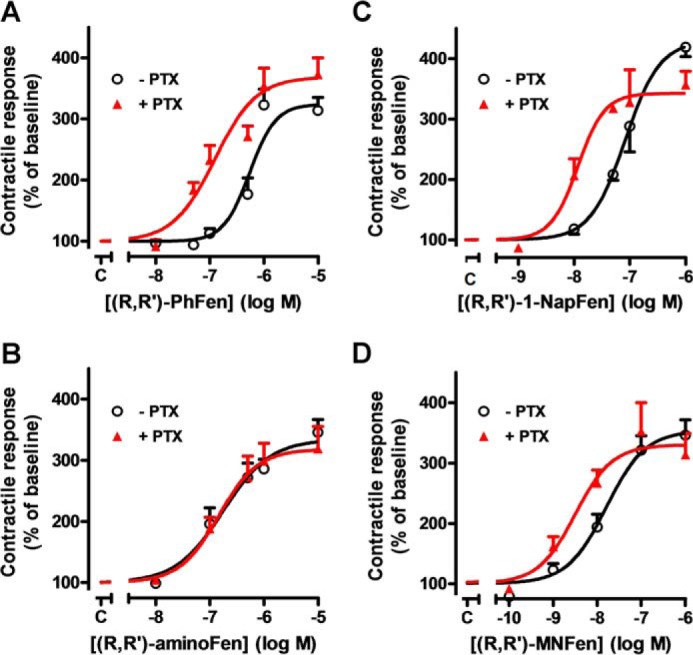
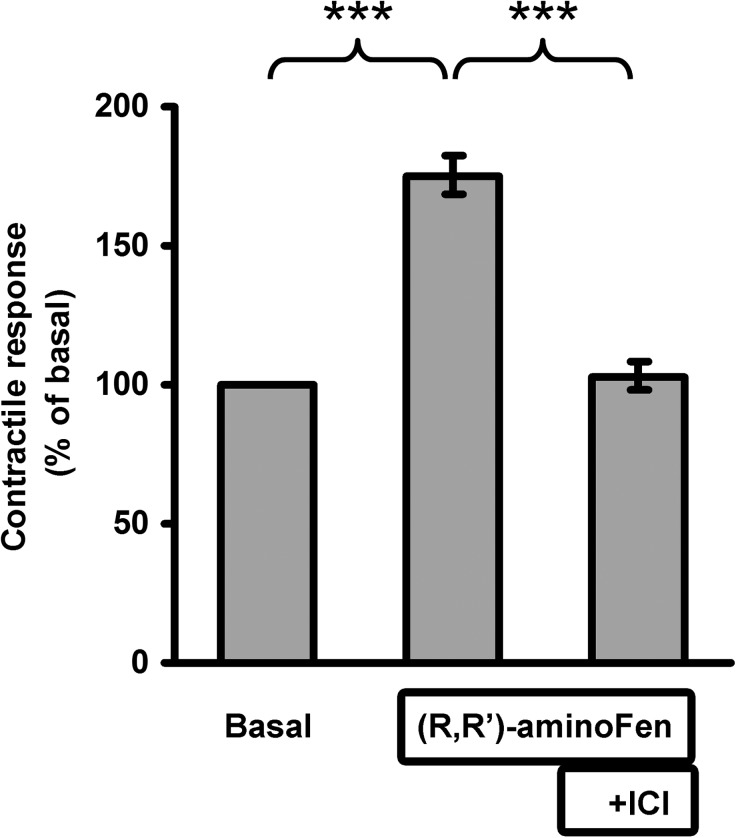
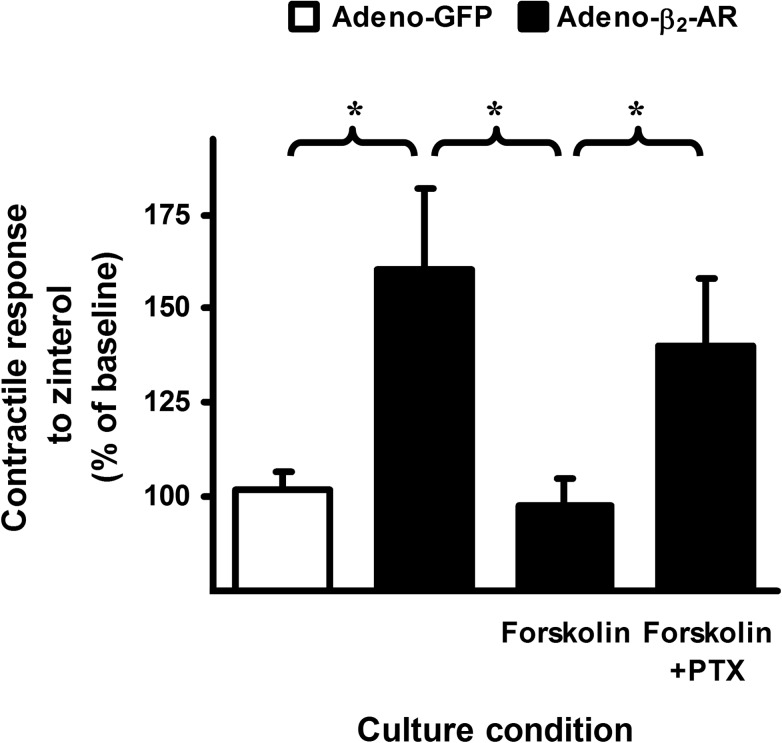
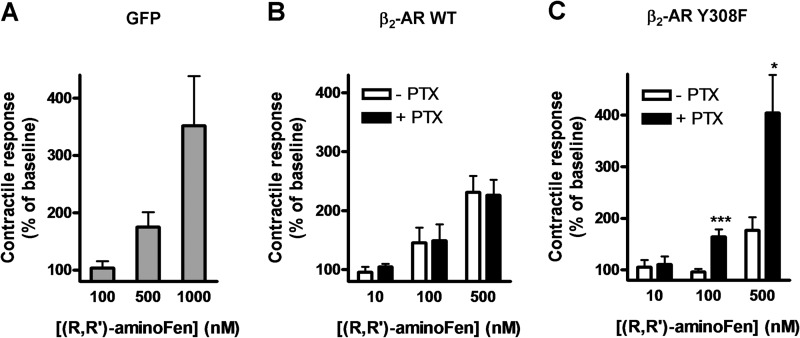
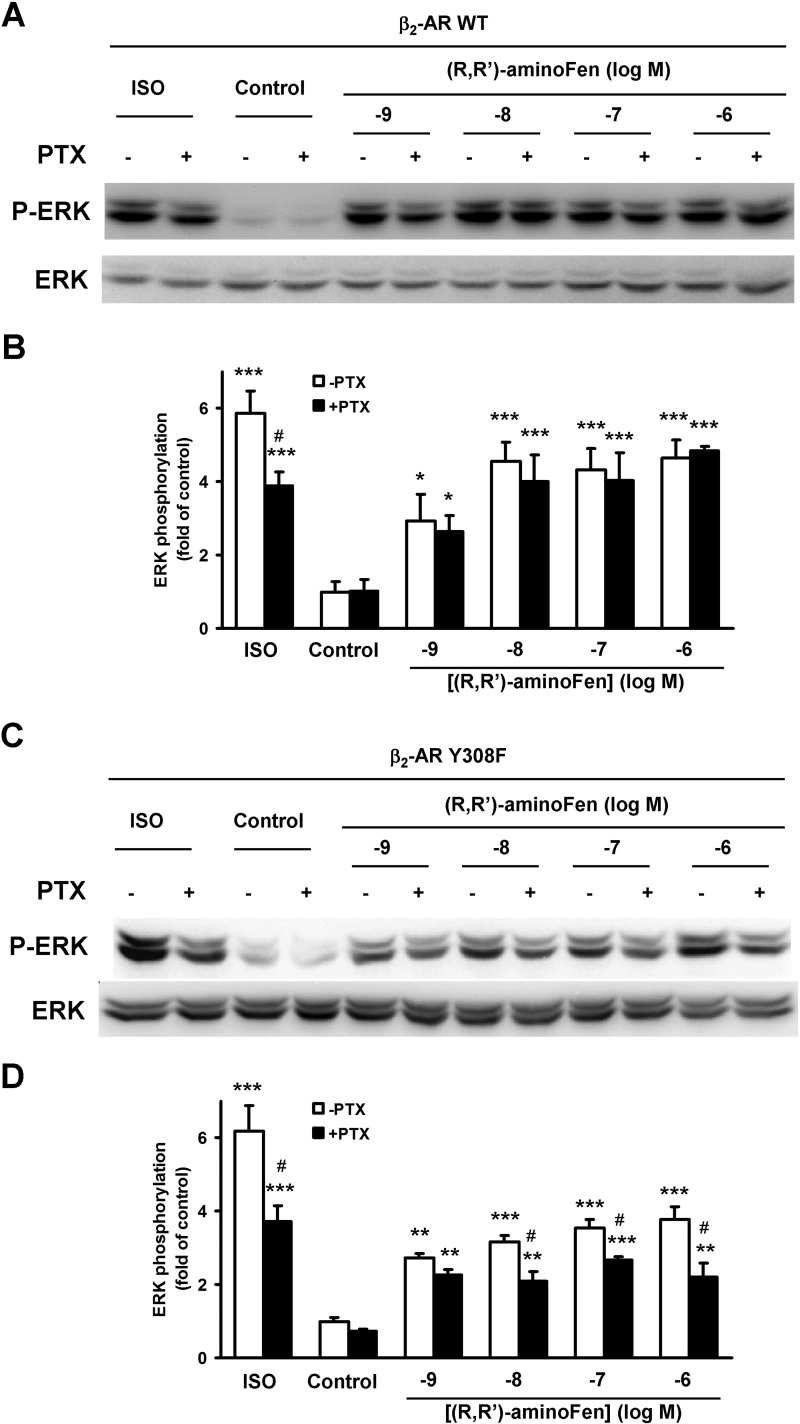
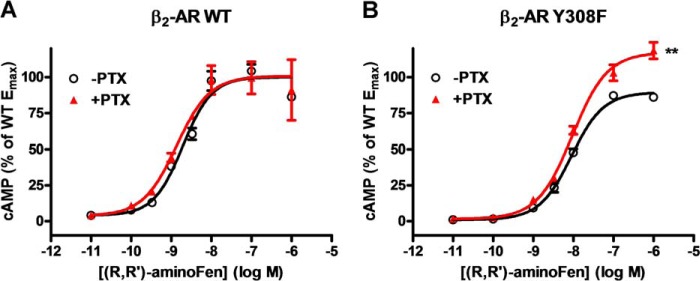
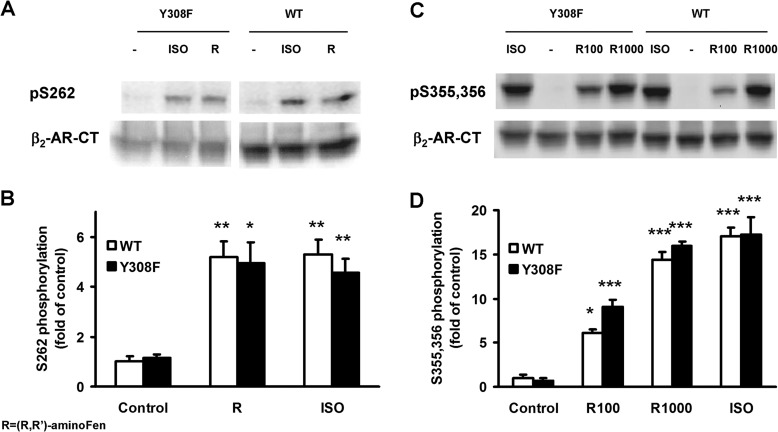
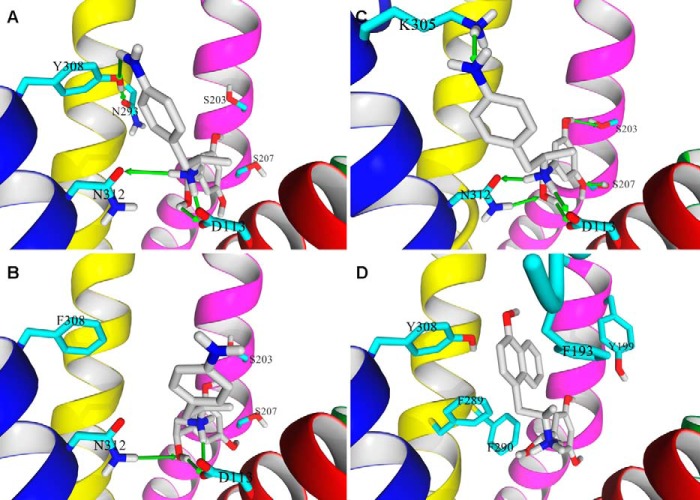
Interaction of a given G protein-coupled receptor to multiple different G proteins is a widespread phenomenon. For instance, β2-adrenoceptor (β2-AR) couples dually to Gs and Gi proteins. Previous studies have shown that cAMP-dependent protein kinase (PKA)-mediated phosphorylation of β2-AR causes a switch in receptor coupling from Gs to Gi. More recent studies have demonstrated that phosphorylation of β2-AR by G protein-coupled receptor kinases, particularly GRK2, markedly enhances the Gi coupling. We have previously shown that although most β2-AR agonists cause both Gs and Gi activation, (R,R')-fenoterol preferentially activates β2-AR-Gs signaling. However, the structural basis for this functional selectivity remains elusive. Here, using docking simulation and site-directed mutagenesis, we defined Tyr-308 as the key amino acid residue on β2-AR essential for Gs-biased signaling. Following stimulation with a β2-AR-Gs-biased agonist (R,R')-4'-aminofenoterol, the Gi disruptor pertussis toxin produced no effects on the receptor-mediated ERK phosphorylation in HEK293 cells nor on the contractile response in cardiomyocytes expressing the wild-type β2-AR. Interestingly, Y308F substitution on β2-AR enabled (R,R')-4'-aminofenoterol to activate Gi and to produce these responses in a pertussis toxin-sensitive manner without altering β2-AR phosphorylation by PKA or G protein-coupled receptor kinases. These results indicate that, in addition to the phosphorylation status, the intrinsic structural feature of β2-AR plays a crucial role in the receptor coupling selectivity to G proteins. We conclude that specific interactions between the ligand and the Tyr-308 residue of β2-AR stabilize receptor conformations favoring the receptor-Gs protein coupling and subsequently result in Gs-biased agonism.
Keywords: Adrenergic Receptor; Cardiomyocyte Contraction; Cardiovascular; Functional Selectivity; G Protein-coupled Receptor (GPCR); Molecular Docking; Molecular Pharmacology; Signal Transduction; Site-directed Mutagenesis.
© 2014 by The American Society for Biochemistry and Molecular Biology, Inc.
Figures
References
-
- Violin J. D., Lefkowitz R. J. (2007) β-Arrestin-biased ligands at seven-transmembrane receptors. Trends Pharmacol. Sci. 28, 416–422 - PubMed
-
- Kenakin T. (2007) Collateral efficacy in drug discovery: taking advantage of the good (allosteric) nature of 7TM receptors. Trends Pharmacol. Sci. 28, 407–415 - PubMed
-
- Urban J. D., Clarke W. P., Zastrow M. V., Nichols D. E., Kobilka B., Weinstein H. (2007) Functional selectivity and classical concepts of quantitative pharmacology. Pharmacology 320, 1–13 - PubMed
-
- Neubig R. R. (2007) Missing links: mechanisms of protean agonism. Mol. Pharmacol. 71, 1200–1202 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
