

Genomic characterization of large heterochromatic gaps in the human genome assembly
- PMID: 24831296
- PMCID: PMC4022460
- DOI: 10.1371/journal.pcbi.1003628
Genomic characterization of large heterochromatic gaps in the human genome assembly
Abstract
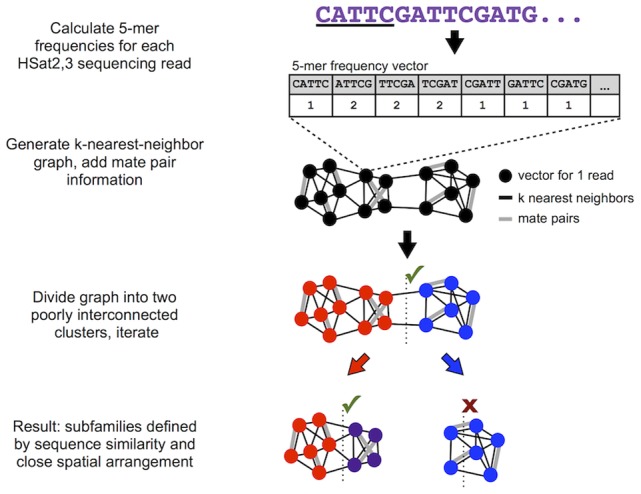
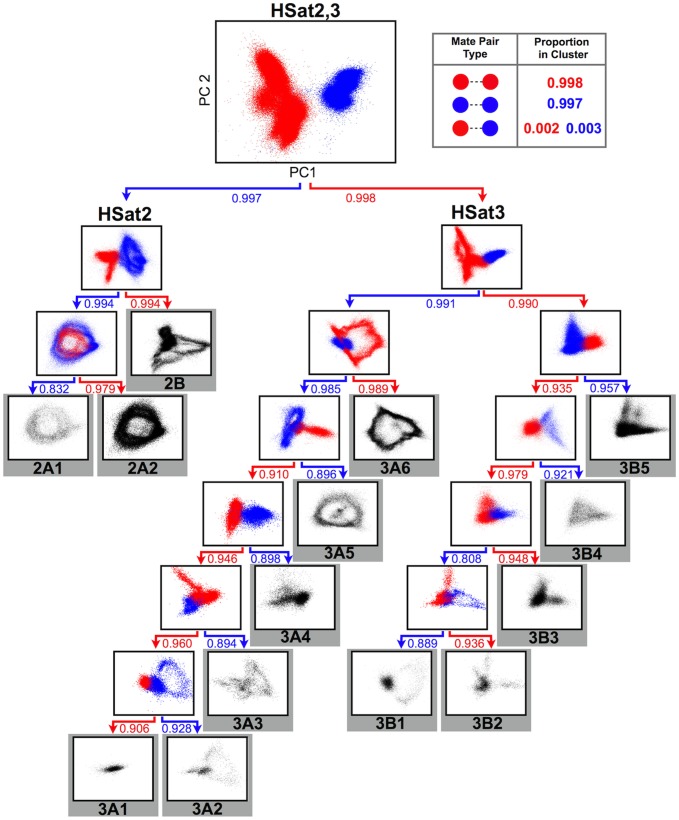
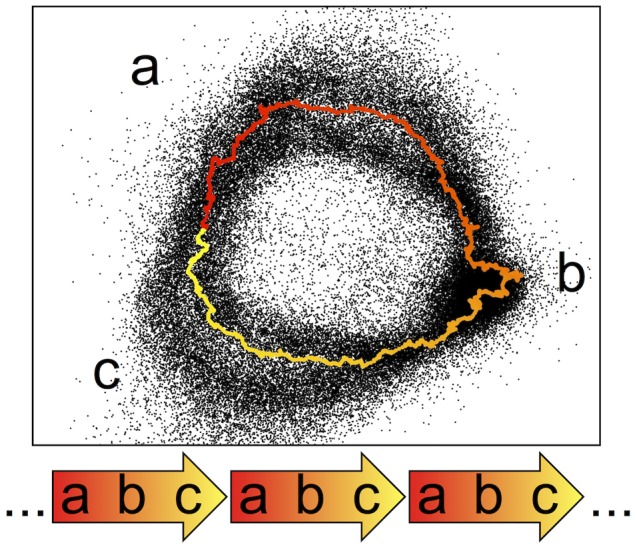
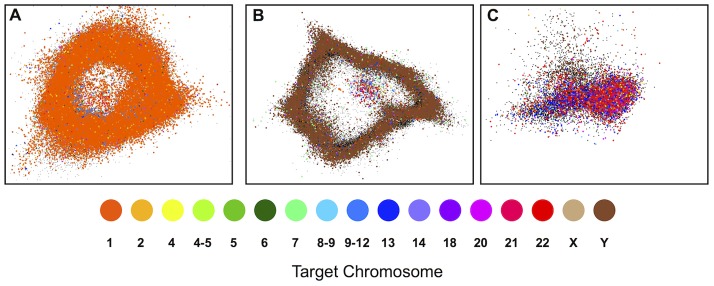
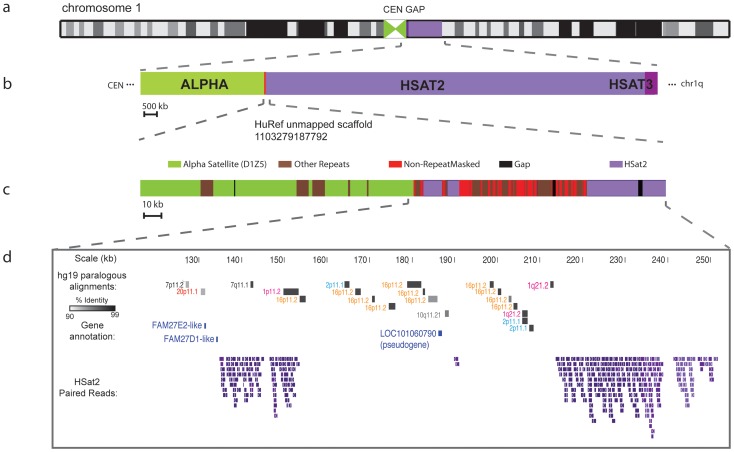
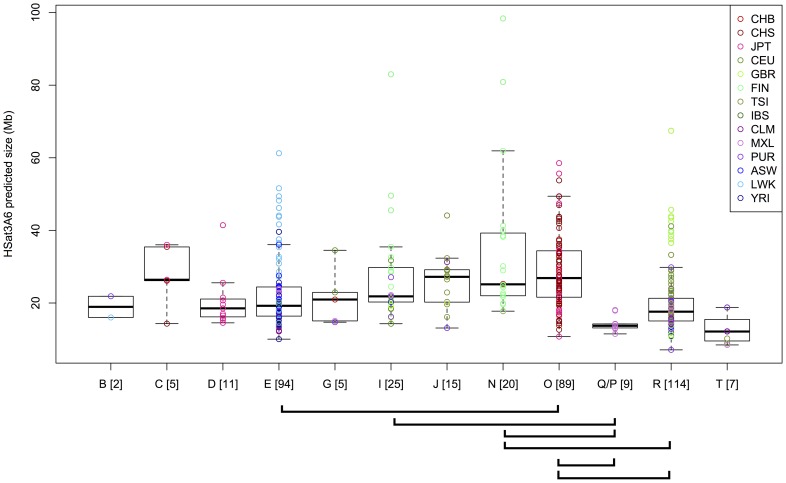
The largest gaps in the human genome assembly correspond to multi-megabase heterochromatic regions composed primarily of two related families of tandem repeats, Human Satellites 2 and 3 (HSat2,3). The abundance of repetitive DNA in these regions challenges standard mapping and assembly algorithms, and as a result, the sequence composition and potential biological functions of these regions remain largely unexplored. Furthermore, existing genomic tools designed to predict consensus-based descriptions of repeat families cannot be readily applied to complex satellite repeats such as HSat2,3, which lack a consistent repeat unit reference sequence. Here we present an alignment-free method to characterize complex satellites using whole-genome shotgun read datasets. Utilizing this approach, we classify HSat2,3 sequences into fourteen subfamilies and predict their chromosomal distributions, resulting in a comprehensive satellite reference database to further enable genomic studies of heterochromatic regions. We also identify 1.3 Mb of non-repetitive sequence interspersed with HSat2,3 across 17 unmapped assembly scaffolds, including eight annotated gene predictions. Finally, we apply our satellite reference database to high-throughput sequence data from 396 males to estimate array size variation of the predominant HSat3 array on the Y chromosome, confirming that satellite array sizes can vary between individuals over an order of magnitude (7 to 98 Mb) and further demonstrating that array sizes are distributed differently within distinct Y haplogroups. In summary, we present a novel framework for generating initial reference databases for unassembled genomic regions enriched with complex satellite DNA, and we further demonstrate the utility of these reference databases for studying patterns of sequence variation within human populations.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
