

Performance comparison of SNP detection tools with illumina exome sequencing data--an assessment using both family pedigree information and sample-matched SNP array data
- PMID: 24831545
- PMCID: PMC4081058
- DOI: 10.1093/nar/gku392
Performance comparison of SNP detection tools with illumina exome sequencing data--an assessment using both family pedigree information and sample-matched SNP array data
Abstract
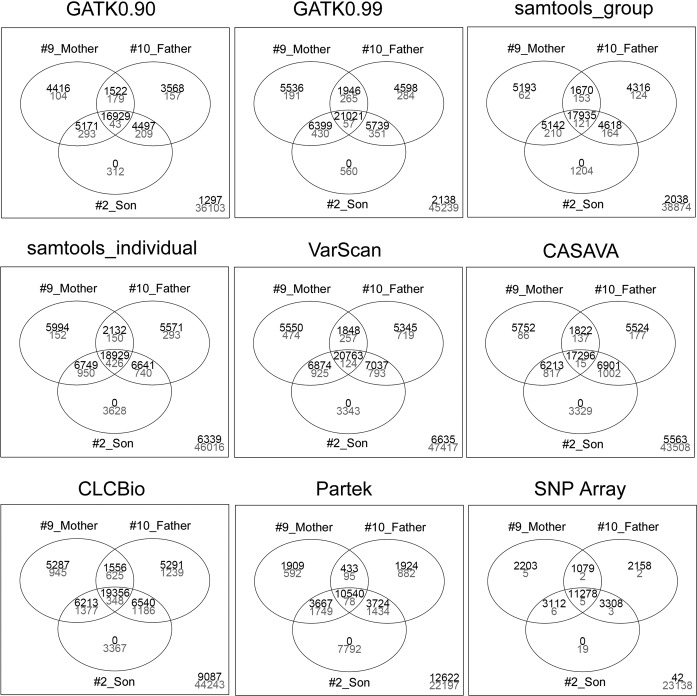
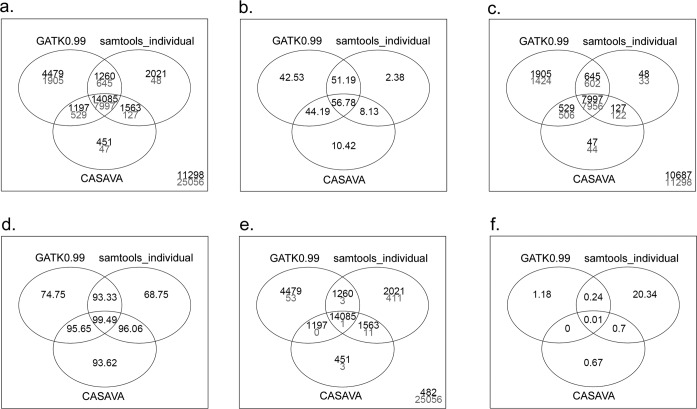
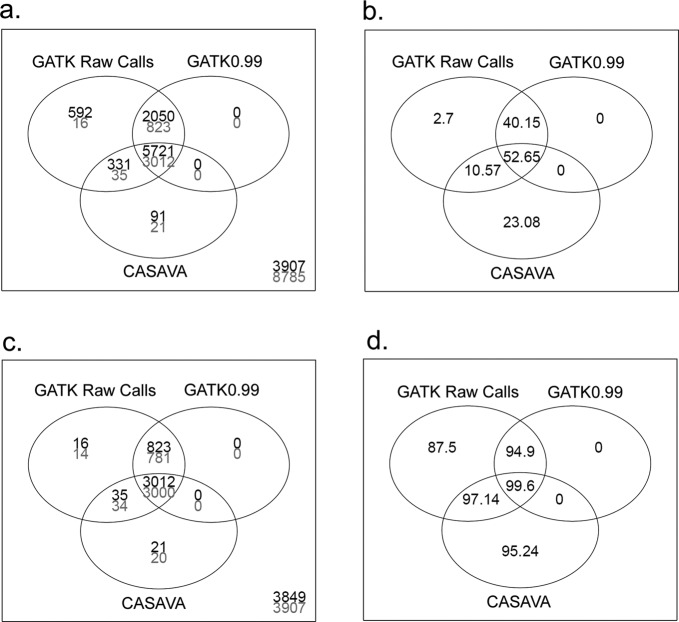
To apply exome-seq-derived variants in the clinical setting, there is an urgent need to identify the best variant caller(s) from a large collection of available options. We have used an Illumina exome-seq dataset as a benchmark, with two validation scenarios--family pedigree information and SNP array data for the same samples, permitting global high-throughput cross-validation, to evaluate the quality of SNP calls derived from several popular variant discovery tools from both the open-source and commercial communities using a set of designated quality metrics. To the best of our knowledge, this is the first large-scale performance comparison of exome-seq variant discovery tools using high-throughput validation with both Mendelian inheritance checking and SNP array data, which allows us to gain insights into the accuracy of SNP calling through such high-throughput validation in an unprecedented way, whereas the previously reported comparison studies have only assessed concordance of these tools without directly assessing the quality of the derived SNPs. More importantly, the main purpose of our study was to establish a reusable procedure that applies high-throughput validation to compare the quality of SNP discovery tools with a focus on exome-seq, which can be used to compare any forthcoming tool(s) of interest.
Published by Oxford University Press on behalf of Nucleic Acids Research 2014. This work is written by (a) US Government employee(s) and is in the public domain in the US.
Figures
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
