

FLEXBAR-Flexible Barcode and Adapter Processing for Next-Generation Sequencing Platforms
- PMID: 24832523
- PMCID: PMC4009805
- DOI: 10.3390/biology1030895
FLEXBAR-Flexible Barcode and Adapter Processing for Next-Generation Sequencing Platforms
Abstract
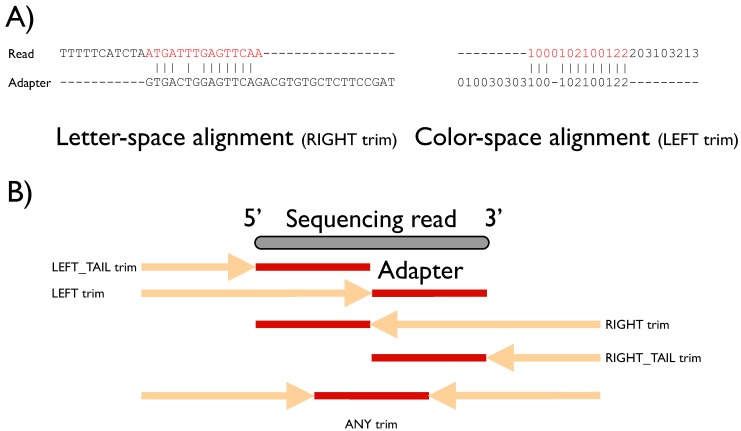
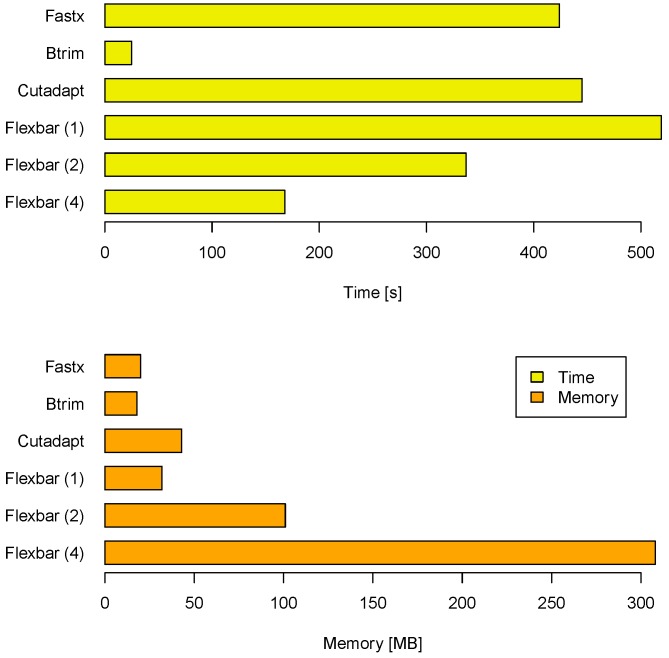
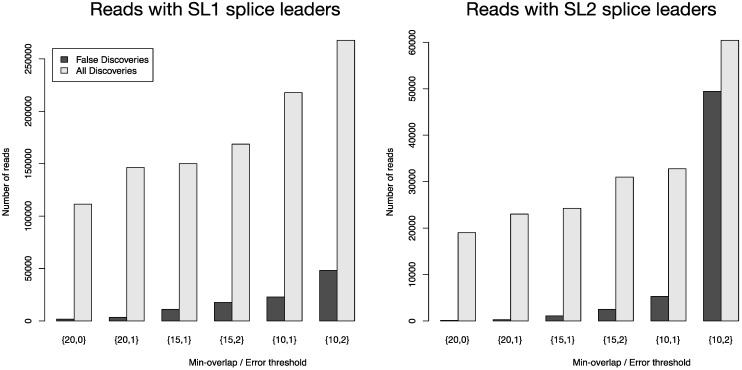
Quantitative and systems biology approaches benefit from the unprecedented depth of next-generation sequencing. A typical experiment yields millions of short reads, which oftentimes carry particular sequence tags. These tags may be: (a) specific to the sequencing platform and library construction method (e.g., adapter sequences); (b) have been introduced by experimental design (e.g., sample barcodes); or (c) constitute some biological signal (e.g., splice leader sequences in nematodes). Our software FLEXBAR enables accurate recognition, sorting and trimming of sequence tags with maximal flexibility, based on exact overlap sequence alignment. The software supports data formats from all current sequencing platforms, including color-space reads. FLEXBAR maintains read pairings and processes separate barcode reads on demand. Our software facilitates the fine-grained adjustment of sequence tag detection parameters and search regions. FLEXBAR is a multi-threaded software and combines speed with precision. Even complex read processing scenarios might be executed with a single command line call. We demonstrate the utility of the software in terms of read mapping applications, library demultiplexing and splice leader detection. FLEXBAR and additional information is available for academic use from the website: http://sourceforge.net/projects/flexbar/.
Figures

References
-
- TBB Library. [(accessed on 14 August 2012)]. Available online: http://www.threadingbuildingblocks.org/
-
- FASTX Toolkit. [(accessed on 25 July 2012)]. Available online: http://hannonlab.cshl.edu/fastx_toolkit/
-
- Martin M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet. J. 2011;17:10–12.
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
