

Die another way--non-apoptotic mechanisms of cell death
- PMID: 24833670
- PMCID: PMC4021468
- DOI: 10.1242/jcs.093575
Die another way--non-apoptotic mechanisms of cell death
Abstract
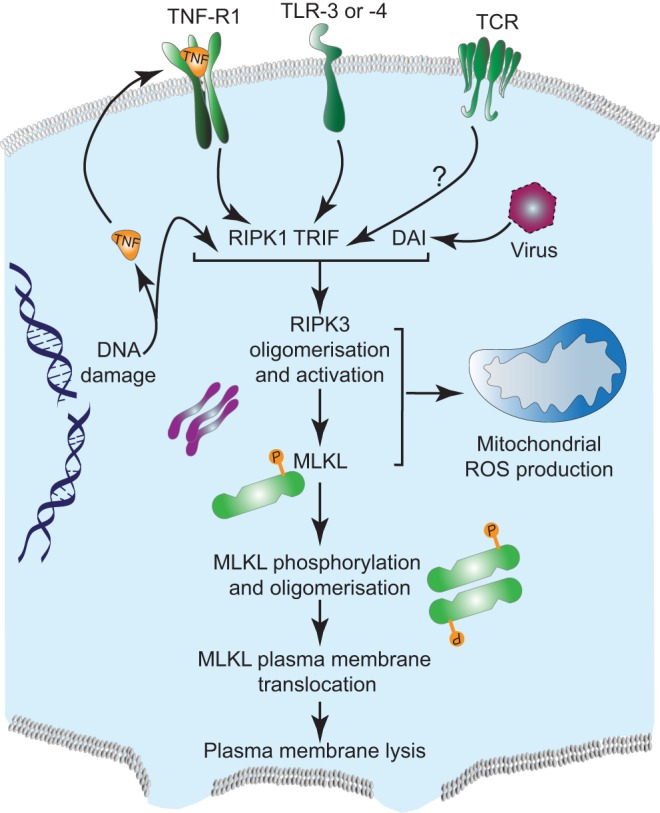
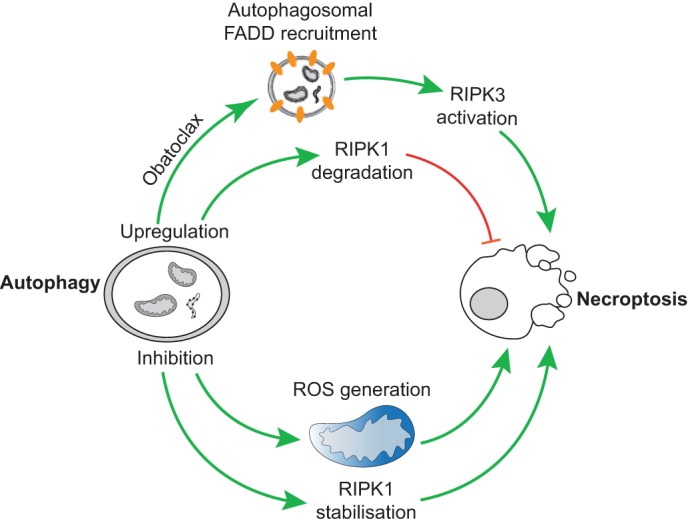
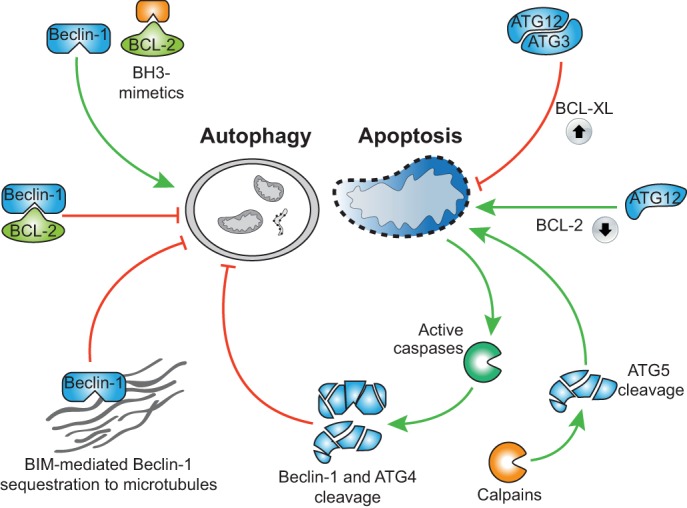
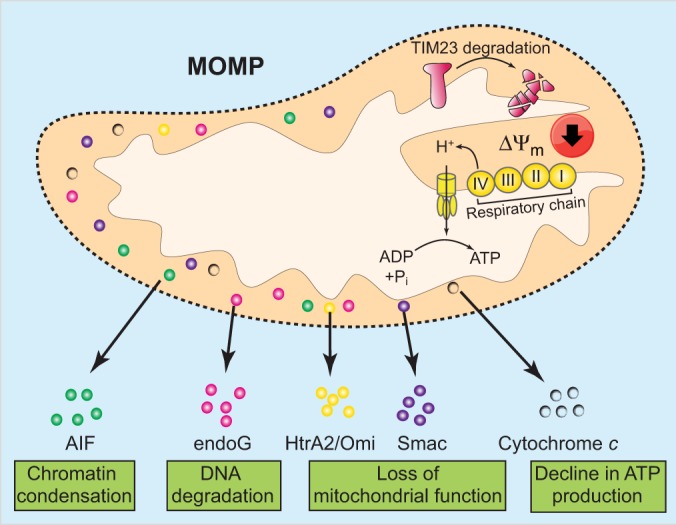
Regulated, programmed cell death is crucial for all multicellular organisms. Cell death is essential in many processes, including tissue sculpting during embryogenesis, development of the immune system and destruction of damaged cells. The best-studied form of programmed cell death is apoptosis, a process that requires activation of caspase proteases. Recently it has been appreciated that various non-apoptotic forms of cell death also exist, such as necroptosis and pyroptosis. These non-apoptotic cell death modalities can be either triggered independently of apoptosis or are engaged should apoptosis fail to execute. In this Commentary, we discuss several regulated non-apoptotic forms of cell death including necroptosis, autophagic cell death, pyroptosis and caspase-independent cell death. We outline what we know about their mechanism, potential roles in vivo and define outstanding questions. Finally, we review data arguing that the means by which a cell dies actually matters, focusing our discussion on inflammatory aspects of cell death.
Keywords: MLKL; Mitochondria; Necroptosis; Pyroptosis; RIPK3.
© 2014. Published by The Company of Biologists Ltd.
Figures
References
-
- Bahi N., Zhang J., Llovera M., Ballester M., Comella J. X., Sanchis D. (2006). Switch from caspase-dependent to caspase-independent death during heart development: essential role of endonuclease G in ischemia-induced DNA processing of differentiated cardiomyocytes. J. Biol. Chem. 281, 22943–22952 10.1074/jbc.M601025200 - DOI - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
