

CaMKII-dependent responses to ischemia and reperfusion challenges in the heart
- PMID: 24834054
- PMCID: PMC4018566
- DOI: 10.3389/fphar.2014.00096
CaMKII-dependent responses to ischemia and reperfusion challenges in the heart
Abstract
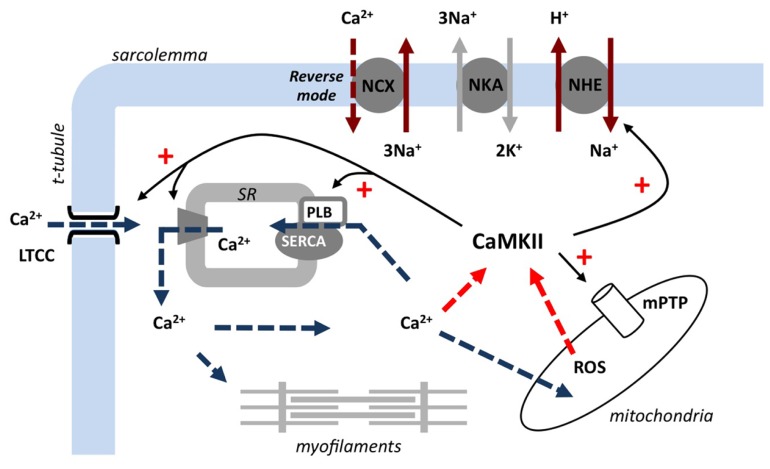
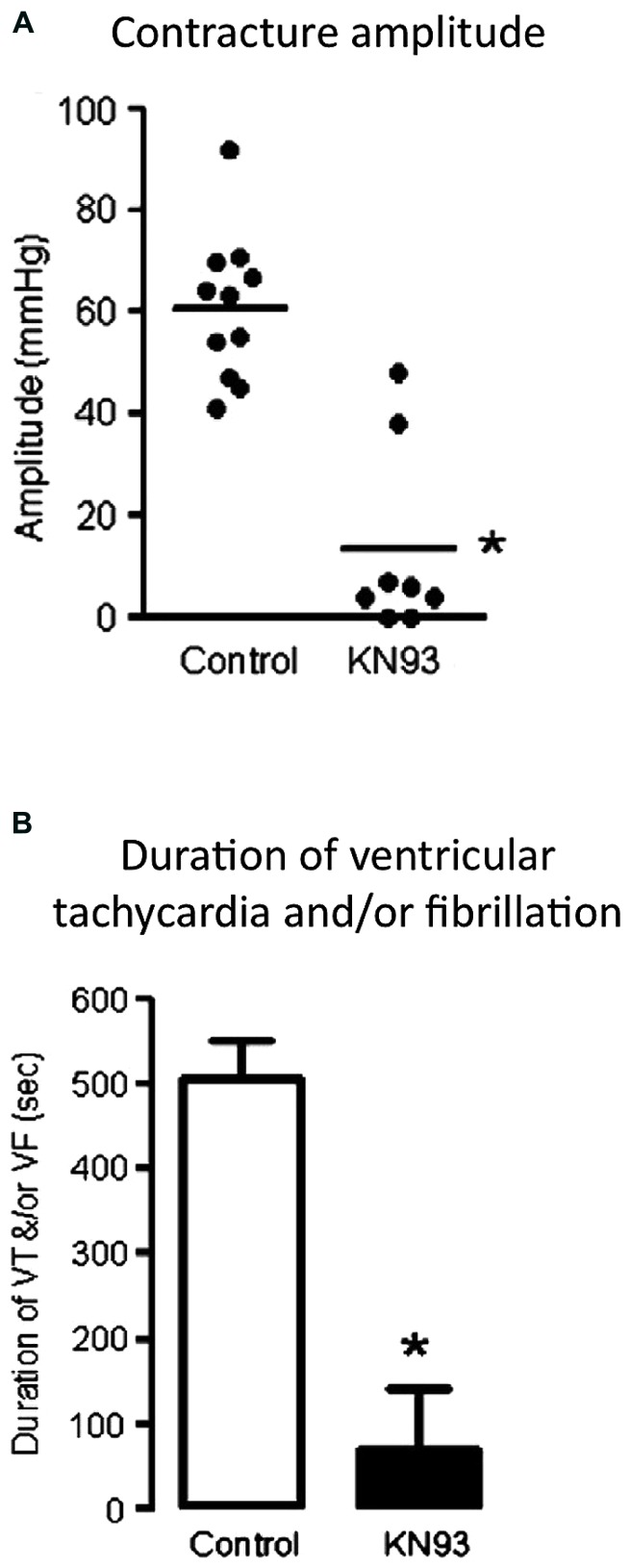
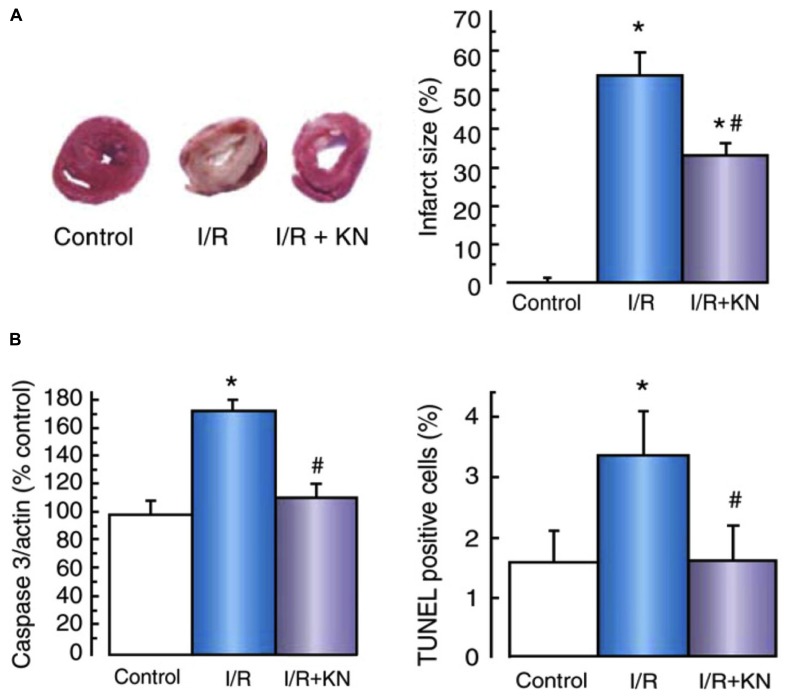
Ischemic heart disease is a leading cause of death, and there is considerable imperative to identify effective therapeutic interventions. Cardiomyocyte Ca(2+) overload is a major cause of ischemia and reperfusion injury, initiating a cascade of events culminating in cardiomyocyte death, myocardial dysfunction, and occurrence of lethal arrhythmias. Responsive to fluctuations in intracellular Ca(2+), Ca(2+)/calmodulin-dependent protein kinase II (CaMKII) has emerged as an enticing therapeutic target in the management of ischemic heart injury. CaMKII is activated early in ischemia and to a greater extent in the first few minutes of reperfusion, at a time when reperfusion arrhythmias are particularly prominent. CaMKII phosphorylates and upregulates many of the key proteins involved in intracellular Na(+) and Ca(2+) loading in ischemia and reperfusion. Experimentally, selective inhibition of CaMKII activity reduces cardiomyocyte death and arrhythmic incidence post-ischemia. New evidence is emerging that CaMKII actions in ischemia and reperfusion involve specific splice variant targeted actions, selective and localized post-translational modifications, and organelle-directed substrate interactions. A more complete mechanistic understanding of CaMKII mode of action in ischemia and reperfusion is required to optimize intervention opportunities. This review summarizes the current experimentally derived understanding of CaMKII participation in mediating the pathophysiology of the heart in ischemia and in reperfusion, and highlights priority future research directions.
Keywords: Ca2+ handling; CaMKII; cardiomyocyte death; contractile function; ischemia; reperfusion.
Figures
Similar articles
-
Ca(2+) /calmodulin dependent kinase II: a critical mediator in determining reperfusion outcomes in the heart?Clin Exp Pharmacol Physiol. 2014 Nov;41(11):940-6. doi: 10.1111/1440-1681.12301. Clin Exp Pharmacol Physiol. 2014. PMID: 25283076 Review.
-
Cardiac CaMKIIδ splice variants exhibit target signaling specificity and confer sex-selective arrhythmogenic actions in the ischemic-reperfused heart.Int J Cardiol. 2015 Feb 15;181:288-96. doi: 10.1016/j.ijcard.2014.11.159. Epub 2014 Nov 26. Int J Cardiol. 2015. PMID: 25539453
-
CaMKIIδ and cardiomyocyte Ca(2+) signalling new perspectives on splice variant targeting.Clin Exp Pharmacol Physiol. 2015 Dec;42(12):1327-32. doi: 10.1111/1440-1681.12489. Clin Exp Pharmacol Physiol. 2015. PMID: 26361740 Review.
-
Novel CaMKII-δ Inhibitor Hesperadin Exerts Dual Functions to Ameliorate Cardiac Ischemia/Reperfusion Injury and Inhibit Tumor Growth.Circulation. 2022 Apr 12;145(15):1154-1168. doi: 10.1161/CIRCULATIONAHA.121.055920. Epub 2022 Mar 23. Circulation. 2022. PMID: 35317609
-
Ca2+/calmodulin-dependent protein kinase inhibition suppresses post-ischemic arrhythmogenesis and mediates sinus bradycardic recovery in reperfusion.Int J Cardiol. 2012 Aug 23;159(2):112-8. doi: 10.1016/j.ijcard.2011.02.038. Epub 2011 Mar 9. Int J Cardiol. 2012. PMID: 21392835
Cited by
-
MicroRNAs and Calcium Signaling in Heart Disease.Int J Mol Sci. 2021 Sep 30;22(19):10582. doi: 10.3390/ijms221910582. Int J Mol Sci. 2021. PMID: 34638924 Free PMC article. Review.
-
CaMKII: The molecular villain that aggravates cardiovascular disease.Exp Ther Med. 2017 Mar;13(3):815-820. doi: 10.3892/etm.2017.4034. Epub 2017 Jan 11. Exp Ther Med. 2017. PMID: 28450904 Free PMC article.
-
Loss of Protein Kinase Novel 1 (PKN1) is associated with mild systolic and diastolic contractile dysfunction, increased phospholamban Thr17 phosphorylation, and exacerbated ischaemia-reperfusion injury.Cardiovasc Res. 2018 Jan 1;114(1):138-157. doi: 10.1093/cvr/cvx206. Cardiovasc Res. 2018. PMID: 29045568 Free PMC article.
-
S-nitrosylation of cardiac myocyte proteins may underlie sex differences in cardiac disease.Front Physiol. 2025 May 6;16:1565917. doi: 10.3389/fphys.2025.1565917. eCollection 2025. Front Physiol. 2025. PMID: 40395646 Free PMC article. Review.
-
New Perspectives on Sex Steroid and Mineralocorticoid Receptor Signaling in Cardiac Ischemic Injury.Front Physiol. 2022 Jun 29;13:896425. doi: 10.3389/fphys.2022.896425. eCollection 2022. Front Physiol. 2022. PMID: 35846011 Free PMC article. Review.
References
-
- Bell J. R., Raaijmakers A. J., Reichelt M. E., Curl C. L., Delbridge L. M. (2013b). Augmented CaMKII recruitment despite less reperfusion arrhythmias in female hearts – a matter of differential post-translational modification of CaMKII splice variants? Proc. Aust. Physiol. Soc. 44:44P (abstract)
-
- Bell J. R., Raaijmakers A. J., Reichelt M. E., Curl C. L., Delbridge L. M. (2014). Differential post-translational modification of CaMKII splice variants may be critical to determining post-ischemic arrhythmia vulnerability in male and female hearts. Global Heart (abstract) in press
-
- Benter I. F., Juggi J. S., Khan I., Yousif M. H., Canatan H., Akhtar S. (2005). Signal transduction mechanisms involved in cardiac preconditioning: role of Ras-GTPase, Ca2+/calmodulin-dependent protein kinase II and epidermal growth factor receptor. Mol. Cell. Biochem. 268 175–183 10.1007/s11010-005-3895-1 - DOI - PubMed
Publication types
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous