

Multiple functional roles of the accessory I-domain of bacteriophage P22 coat protein revealed by NMR structure and CryoEM modeling
- PMID: 24836025
- PMCID: PMC4068711
- DOI: 10.1016/j.str.2014.04.003
Multiple functional roles of the accessory I-domain of bacteriophage P22 coat protein revealed by NMR structure and CryoEM modeling
Abstract
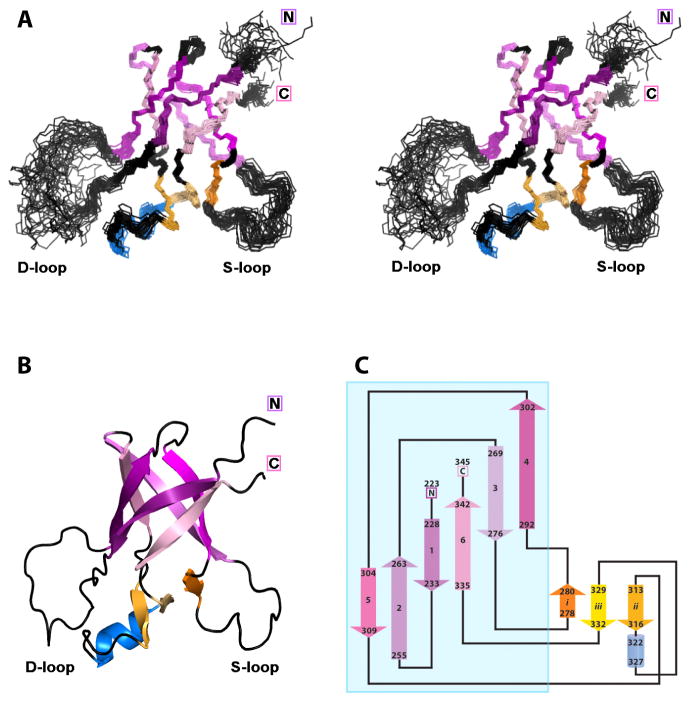
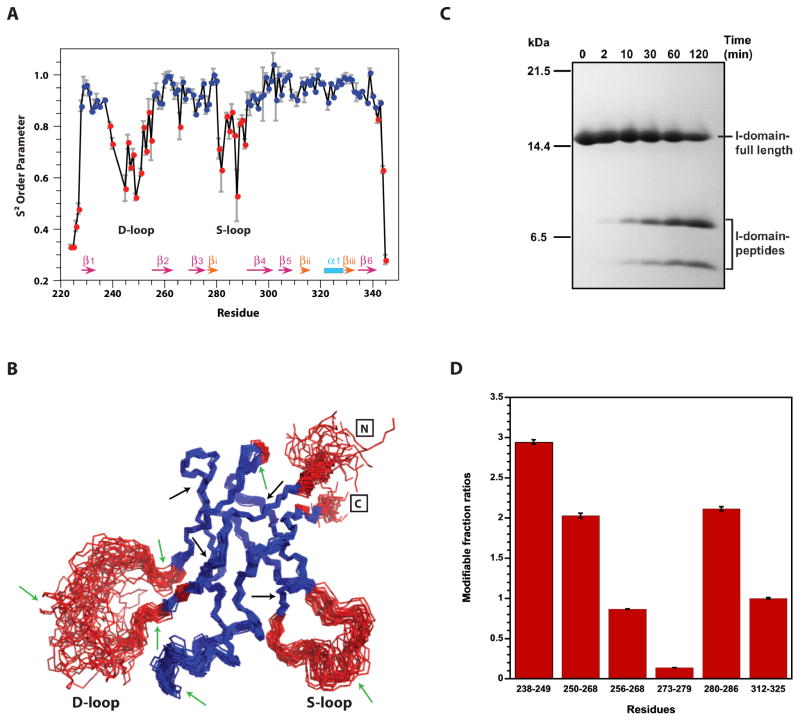
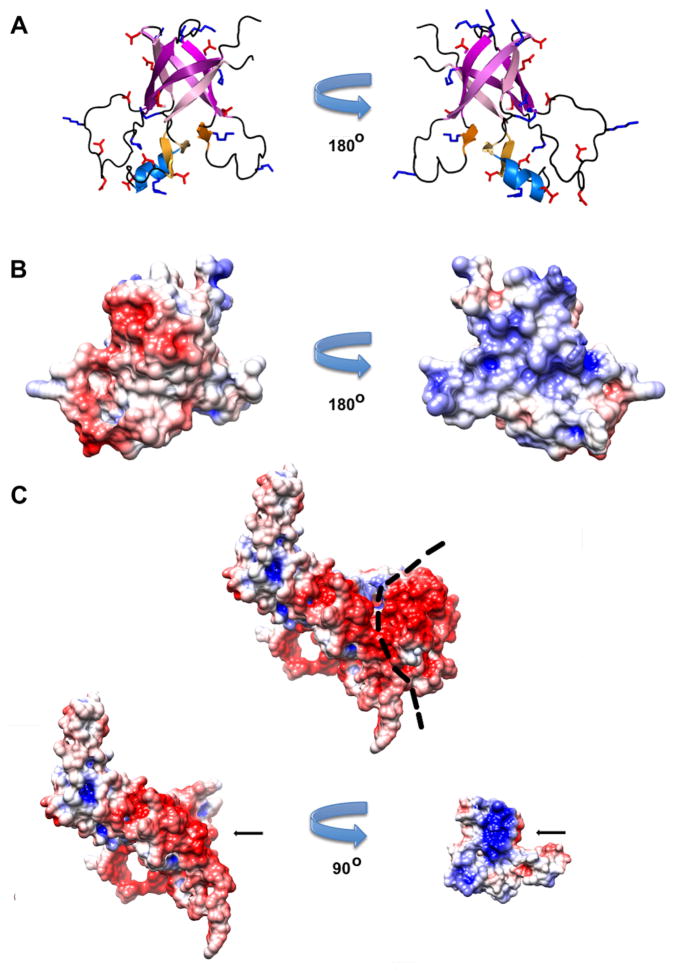
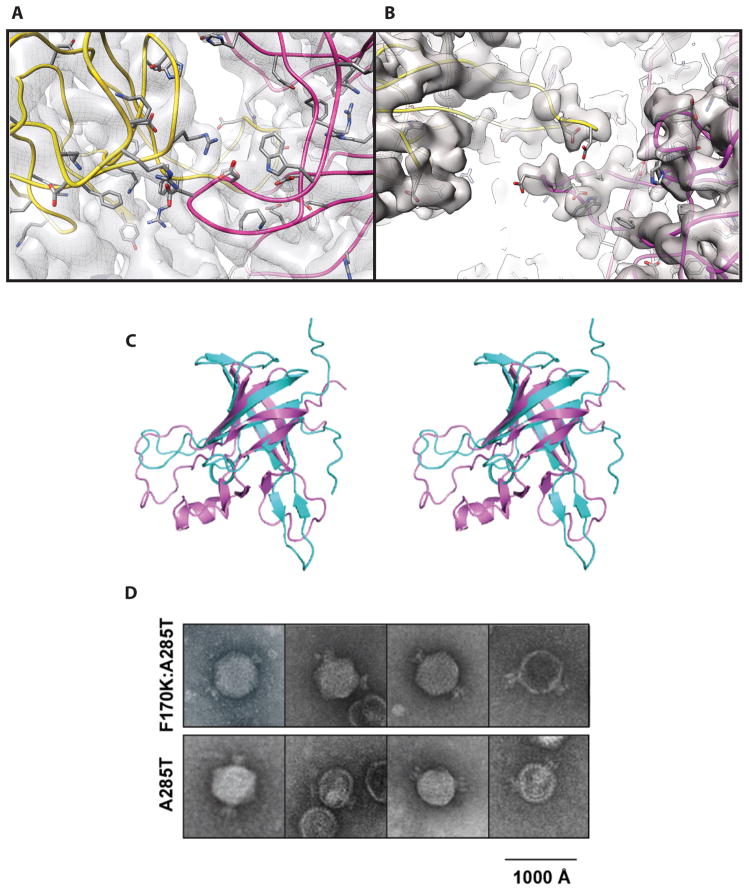
Some capsid proteins built on the ubiquitous HK97-fold have accessory domains imparting specific functions. Bacteriophage P22 coat protein has a unique insertion domain (I-domain). Two prior I-domain models from subnanometer cryoelectron microscopy (cryoEM) reconstructions differed substantially. Therefore, the I-domain's nuclear magnetic resonance structure was determined and also used to improve cryoEM models of coat protein. The I-domain has an antiparallel six-stranded β-barrel fold, not previously observed in HK97-fold accessory domains. The D-loop, which is dynamic in the isolated I-domain and intact monomeric coat protein, forms stabilizing salt bridges between adjacent capsomers in procapsids. The S-loop is important for capsid size determination, likely through intrasubunit interactions. Ten of 18 coat protein temperature-sensitive-folding substitutions are in the I-domain, indicating its importance in folding and stability. Several are found on a positively charged face of the β-barrel that anchors the I-domain to a negatively charged surface of the coat protein HK97-core.
Copyright © 2014 Elsevier Ltd. All rights reserved.
Figures


References
-
- Abrescia NG, Bamford DH, Grimes JM, Stuart DI. Structure unifies the viral universe. Annual Review of Biochemistry. 2012;81:795–822. - PubMed
-
- Akita F, Chong KT, Tanaka H, Yamashita E, Miyazaki N, Nakaishi Y, Suzuki M, Namba K, Ono Y, Tsukihara T, et al. The crystal structure of a virus-like particle from the hyperthermophilic archaeon Pyrococcus furiosus provides insight into the evolution of viruses. Journal of Molecular Biology. 2007;368:1469–1483. - PubMed
-
- Alexandrescu AT, Jahnke W, Wiltscheck R, Blommers MJ. Accretion of structure in staphylococcal nuclease: an 15N NMR relaxation study. Journal of molecular biology. 1996;260:570–587. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
