

Whole-exome sequencing and clinical interpretation of formalin-fixed, paraffin-embedded tumor samples to guide precision cancer medicine
- PMID: 24836576
- PMCID: PMC4048335
- DOI: 10.1038/nm.3559
Whole-exome sequencing and clinical interpretation of formalin-fixed, paraffin-embedded tumor samples to guide precision cancer medicine
Abstract
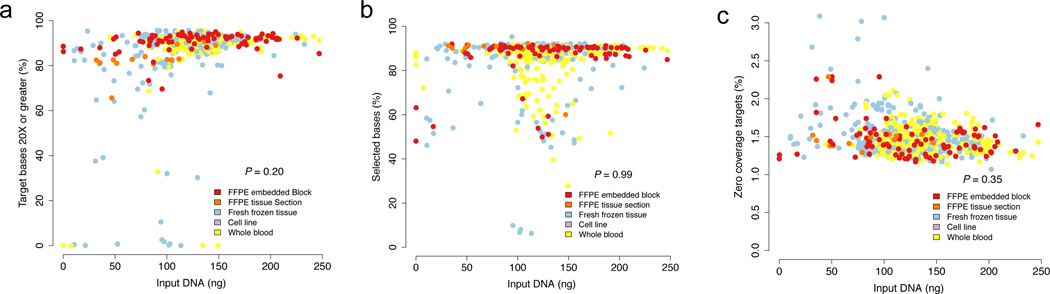
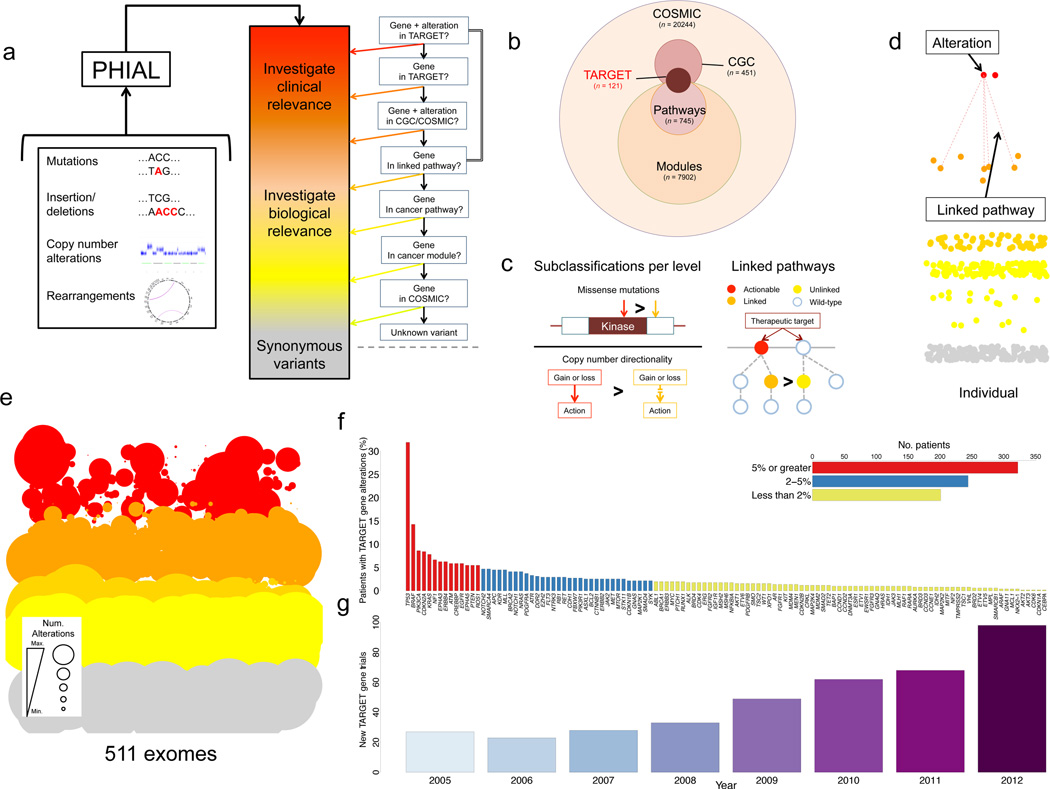
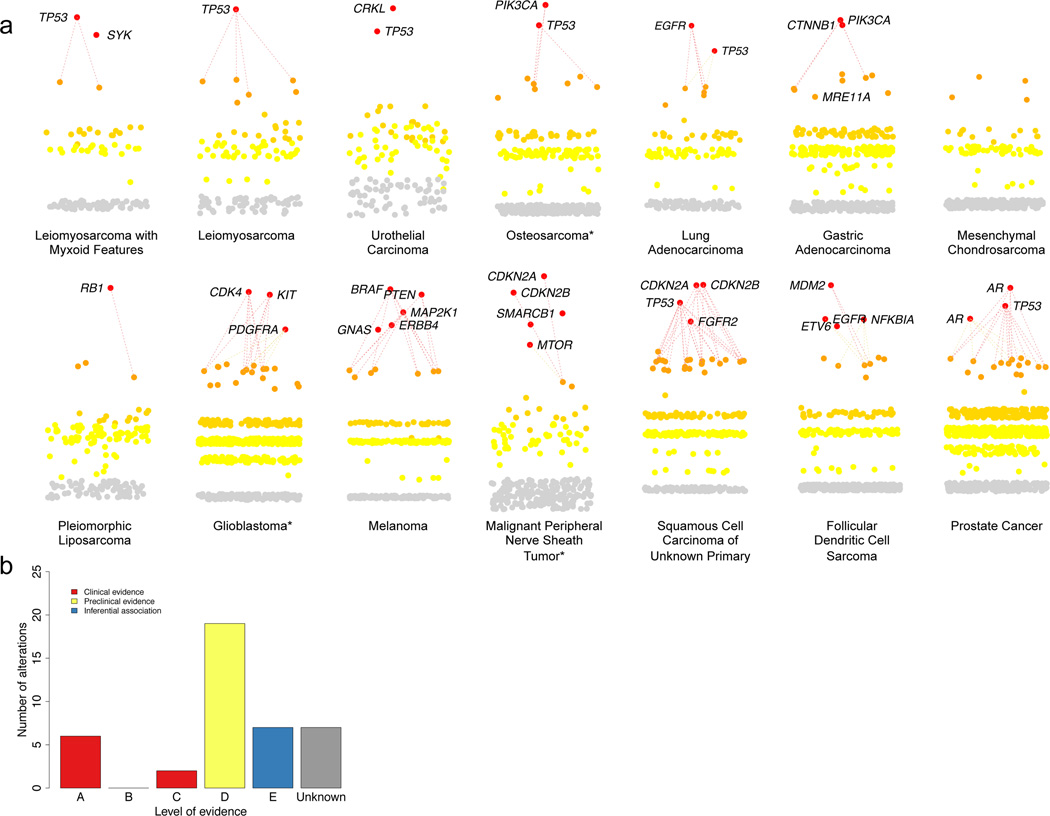
Translating whole-exome sequencing (WES) for prospective clinical use may have an impact on the care of patients with cancer; however, multiple innovations are necessary for clinical implementation. These include rapid and robust WES of DNA derived from formalin-fixed, paraffin-embedded tumor tissue, analytical output similar to data from frozen samples and clinical interpretation of WES data for prospective use. Here, we describe a prospective clinical WES platform for archival formalin-fixed, paraffin-embedded tumor samples. The platform employs computational methods for effective clinical analysis and interpretation of WES data. When applied retrospectively to 511 exomes, the interpretative framework revealed a 'long tail' of somatic alterations in clinically important genes. Prospective application of this approach identified clinically relevant alterations in 15 out of 16 patients. In one patient, previously undetected findings guided clinical trial enrollment, leading to an objective clinical response. Overall, this methodology may inform the widespread implementation of precision cancer medicine.
Figures


References
-
- Garraway LA, Lander ES. Lessons from the cancer genome. Cell. 2013;153:17–37. - PubMed
-
- Garraway LA. Genomics-Driven Oncology: Framework for an Emerging Paradigm. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2013 - PubMed
-
- Garraway LA, Janne PA. Circumventing cancer drug resistance in the era of personalized medicine. Cancer discovery. 2012;2:214–226. - PubMed
-
- Thomas RK, Baker AC, Debiasi RM, Winckler W, Laframboise T, et al. High-throughput oncogene mutation profiling in human cancer. Nature genetics. 2007;39:347–351. - PubMed
Online Methods References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
