

Cardiac BIN1 folds T-tubule membrane, controlling ion flux and limiting arrhythmia
- PMID: 24836577
- PMCID: PMC4048325
- DOI: 10.1038/nm.3543
Cardiac BIN1 folds T-tubule membrane, controlling ion flux and limiting arrhythmia
Abstract
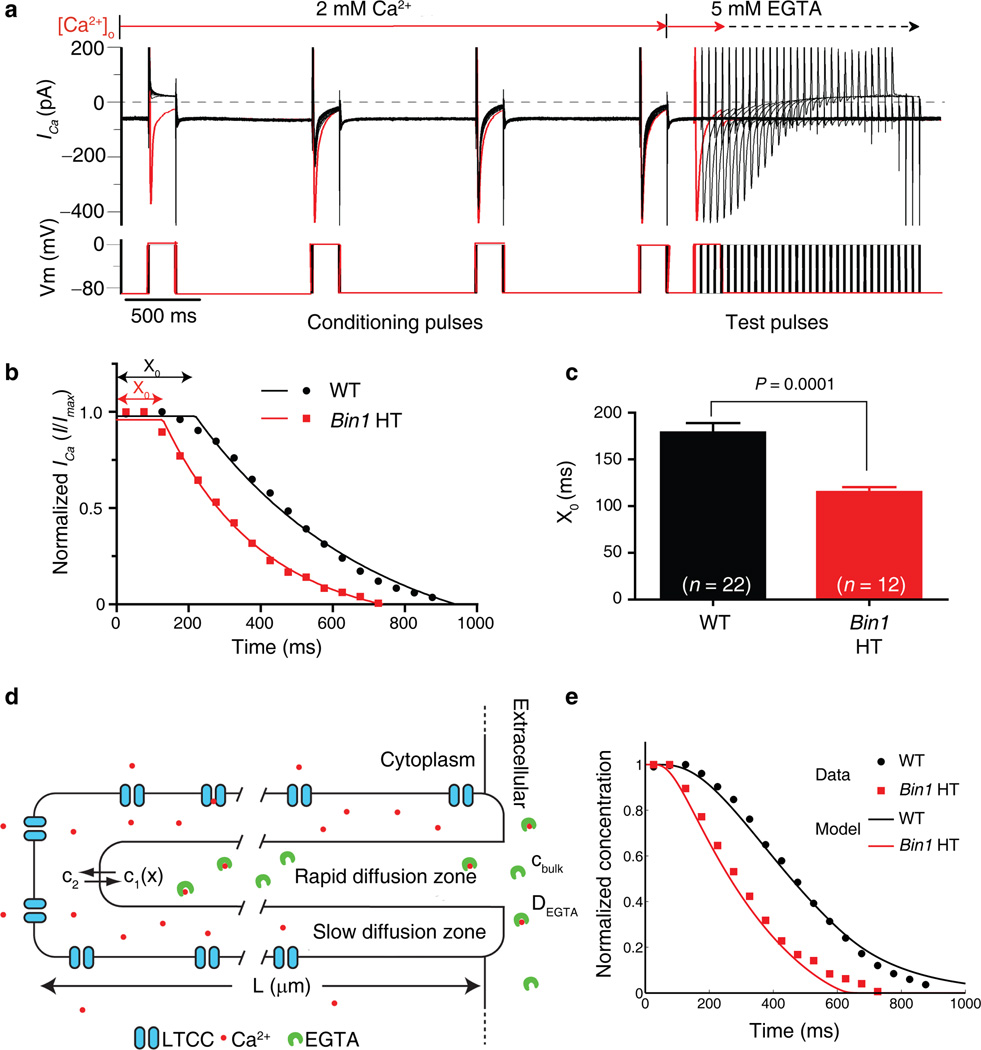
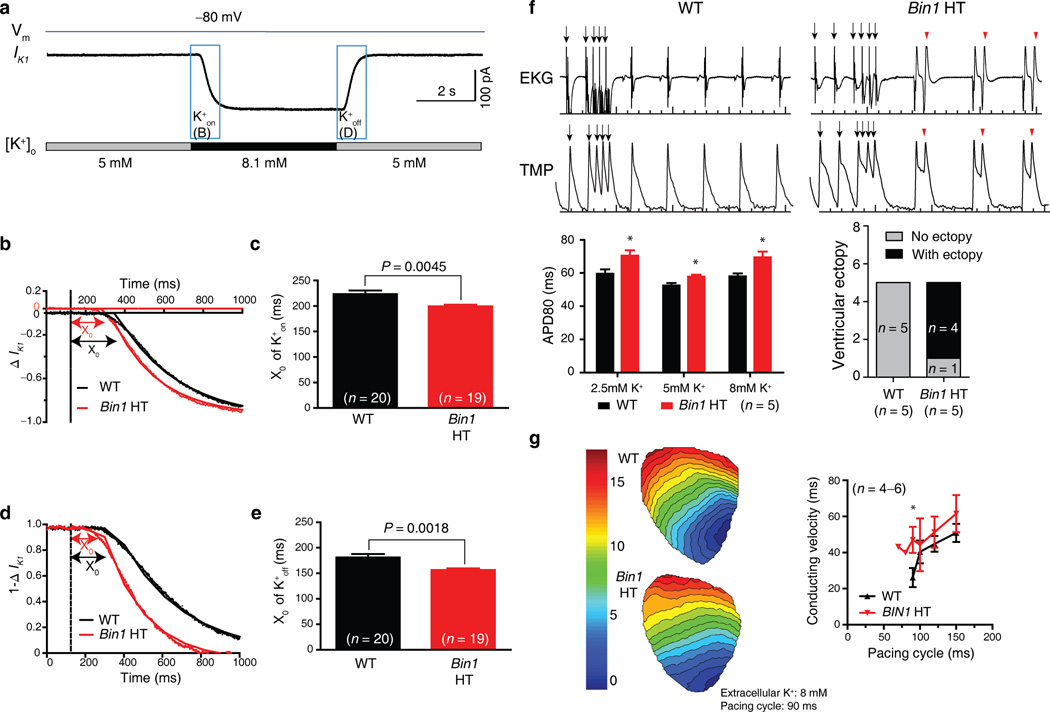
Cardiomyocyte T tubules are important for regulating ion flux. Bridging integrator 1 (BIN1) is a T-tubule protein associated with calcium channel trafficking that is downregulated in failing hearts. Here we find that cardiac T tubules normally contain dense protective inner membrane folds that are formed by a cardiac isoform of BIN1. In mice with cardiac Bin1 deletion, T-tubule folding is decreased, which does not change overall cardiomyocyte morphology but leads to free diffusion of local extracellular calcium and potassium ions, prolonging action-potential duration and increasing susceptibility to ventricular arrhythmias. We also found that T-tubule inner folds are rescued by expression of the BIN1 isoform BIN1+13+17, which promotes N-WASP-dependent actin polymerization to stabilize the T-tubule membrane at cardiac Z discs. BIN1+13+17 recruits actin to fold the T-tubule membrane, creating a 'fuzzy space' that protectively restricts ion flux. When the amount of the BIN1+13+17 isoform is decreased, as occurs in acquired cardiomyopathy, T-tubule morphology is altered, and arrhythmia can result.
Figures




References
-
- Soeller C, Cannell MB. Examination of the transverse tubular system in living cardiac rat myocytes by 2-photon microscopy and digital image-processing techniques. Circ Res. 1999;84:266–275. - PubMed
-
- Brette F, Orchard C. T-tubule function in mammalian cardiac myocytes. Circ Res. 2003;92:1182–1192. - PubMed
-
- Cheng H, Lederer WJ, Cannell MB. Calcium sparks: elementary events underlying excitation-contraction coupling in heart muscle. Science. 1993;262:740–744. - PubMed
-
- Bers DM. Cardiac excitation-contraction coupling. Nature. 2002;415:198–205. - PubMed
-
- Pasek M, Simurda J, Christe G. The functional role of cardiac T-tubules explored in a model of rat ventricular myocytes. Philos Transact A Math Phys Eng Sci. 2006;364:1187–1206. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
