

Identification of two novel mutations in the PHEX gene in Chinese patients with hypophosphatemic rickets/osteomalacia
- PMID: 24836714
- PMCID: PMC4024000
- DOI: 10.1371/journal.pone.0097830
Identification of two novel mutations in the PHEX gene in Chinese patients with hypophosphatemic rickets/osteomalacia
Abstract
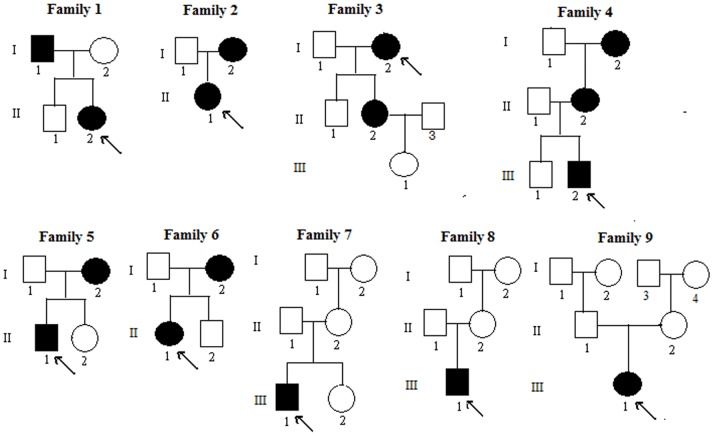
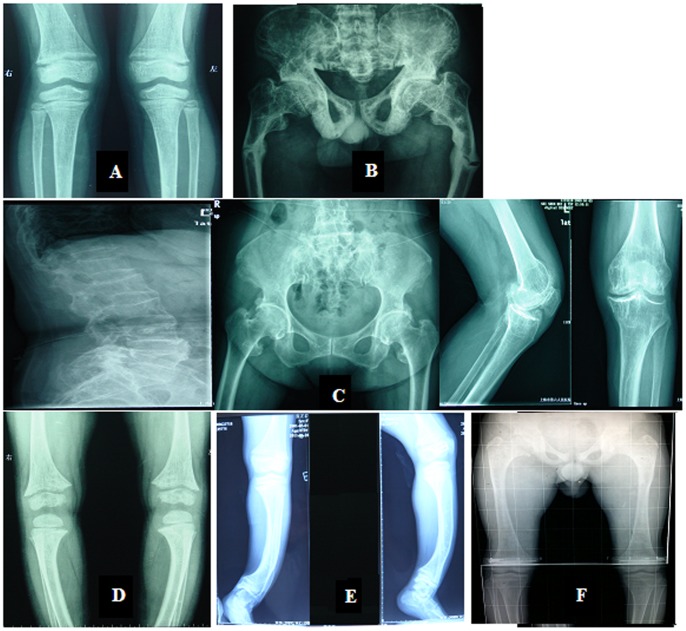
Objective: X-linked dominant hypophosphatemia (XLH) is the most prevalent form of inherited rickets/osteomalacia in humans. The aim of this study was to identify PHEX gene mutations and describe the clinical features observed in 6 unrelated Chinese families and 3 sporadic patients with hypophosphatemic rickets/osteomalacia.
Methods: For this study, 45 individuals from 9 unrelated families of Chinese Han ethnicity (including 16 patients and 29 normal phenotype subjects), and 250 healthy donors were recruited. All 22 exons and exon-intron boundaries of the PHEX gene were amplified by polymerase chain reaction (PCR) and directly sequenced.
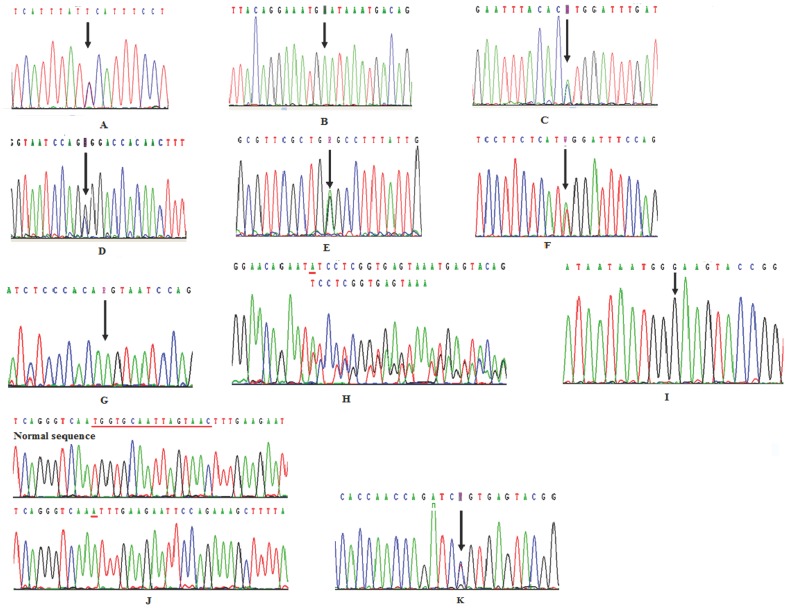
Results: The PHEX mutations were detected in 6 familial and 3 sporadic hypophosphatemic rickets/osteomalacia. Altogether, 2 novel mutations were detected: 1 missense mutation c.1183G>C in exon 11, resulting in p.Gly395Arg and 1 missense mutation c.1751A>C in exon 17, resulting in p.His584Pro. No mutations were found in the 250 healthy controls.
Conclusions: Our study increases knowledge of the PHEX gene mutation types and clinical phenotypes found in Chinese patients with XLH, which is important for understanding the genetic basis of XLH. The molecular diagnosis of a PHEX genetic mutation is of great importance for confirming the clinical diagnosis of XLH, conducting genetic counseling, and facilitating prenatal intervention, especially in the case of sporadic patients.
Conflict of interest statement
Figures

Similar articles
-
Seven novel and six de novo PHEX gene mutations in patients with hypophosphatemic rickets.Int J Mol Med. 2016 Dec;38(6):1703-1714. doi: 10.3892/ijmm.2016.2796. Epub 2016 Nov 7. Int J Mol Med. 2016. PMID: 27840894 Free PMC article.
-
PHEX 3'-UTR c.*231A>G near the polyadenylation signal is a relatively common, mild, American mutation that masquerades as sporadic or X-linked recessive hypophosphatemic rickets.J Bone Miner Res. 2015 Jan;30(1):137-43. doi: 10.1002/jbmr.2307. J Bone Miner Res. 2015. PMID: 25042154 Clinical Trial.
-
Three novel PHEX gene mutations in four Chinese families with X-linked dominant hypophosphatemic rickets.Biochem Biophys Res Commun. 2012 Jul 13;423(4):793-8. doi: 10.1016/j.bbrc.2012.06.042. Epub 2012 Jun 16. Biochem Biophys Res Commun. 2012. PMID: 22713460
-
PHEX gene mutation in a Chinese family with six cases of X-linked hypophosphatemic rickets.J Pediatr Endocrinol Metab. 2013;26(11-12):1179-83. doi: 10.1515/jpem-2013-0101. J Pediatr Endocrinol Metab. 2013. PMID: 23813354 Review.
-
A mosaic mutation of phosphate-regulating gene with homologies to endopeptidases on the X chromosome (PHEX) in X-linked hypophosphatemic rickets with mild bone phenotypes.Endocr J. 2021 Sep 28;68(9):1135-1141. doi: 10.1507/endocrj.EJ20-0809. Epub 2021 Apr 28. Endocr J. 2021. PMID: 33907069 Review.
Cited by
-
Asia-Pacific Consensus Recommendations on X-Linked Hypophosphatemia: Diagnosis, Multidisciplinary Management, and Transition From Pediatric to Adult Care.JBMR Plus. 2023 May 1;7(6):e10744. doi: 10.1002/jbm4.10744. eCollection 2023 Jun. JBMR Plus. 2023. PMID: 37283655 Free PMC article. Review.
-
Genetic Testing Confirmed the Early Diagnosis of X-Linked Hypophosphatemic Rickets in a 7-Month-Old Infant.J Investig Med High Impact Case Rep. 2015 Aug 3;3(3):2324709615598167. doi: 10.1177/2324709615598167. eCollection 2015 Jul-Sep. J Investig Med High Impact Case Rep. 2015. PMID: 26904698 Free PMC article.
-
Clinical and Genetic Characteristics of 153 Chinese Patients With X-Linked Hypophosphatemia.Front Cell Dev Biol. 2021 Jun 1;9:617738. doi: 10.3389/fcell.2021.617738. eCollection 2021. Front Cell Dev Biol. 2021. PMID: 34141703 Free PMC article.
-
A novel c.2179T>C mutation blocked the intracellular transport of PHEX protein and caused X-linked hypophosphatemic rickets in a Chinese family.Mol Genet Genomic Med. 2020 Aug;8(8):e1262. doi: 10.1002/mgg3.1262. Epub 2020 Jun 8. Mol Genet Genomic Med. 2020. PMID: 32511895 Free PMC article.
-
Whole Exome Sequencing Reveals Novel PHEX Splice Site Mutations in Patients with Hypophosphatemic Rickets.PLoS One. 2015 Jun 24;10(6):e0130729. doi: 10.1371/journal.pone.0130729. eCollection 2015. PLoS One. 2015. PMID: 26107949 Free PMC article.
References
-
- Rowe PS (1994) Molecular biology of hypophosphataemic rickets and oncogenic osteomalacia. Hum Genet 94: 457–467. - PubMed
-
- Rowe PS, Oudet CL, Francis F, Sinding C, Pannetier S, et al. (1997) Distribution of mutations in the PHEX gene in families with X-linked hypophosphataemic rickets (HYP). Hum Mol Genet 6: 539–549. - PubMed
-
- Rowe PS (1998) The role of the PHEX gene (PEX) in families with X-linked hypophosphataemic rickets. Curr Opin Nephrol Hypertens 7: 367–376. - PubMed
-
- Quarles LD, Drezner MK (2001) Pathophysiology of X-linked hypophosphatemia, tumor-induced osteomalacia, and autosomal dominant hypophosphatemia: a perPHEXing problem. J Clin Endocrinol Metab 86: 494–496. - PubMed
-
- Albright F, Butler A, Bloomberg E (1939) Rickets resistant to vitamin D therapy. American Journal of Disease of Children 54: 529–547.
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
