

Pivotal role of MUC1 glycosylation by cigarette smoke in modulating disruption of airway adherens junctions in vitro
- PMID: 24838315
- PMCID: PMC4138268
- DOI: 10.1002/path.4375
Pivotal role of MUC1 glycosylation by cigarette smoke in modulating disruption of airway adherens junctions in vitro
Abstract
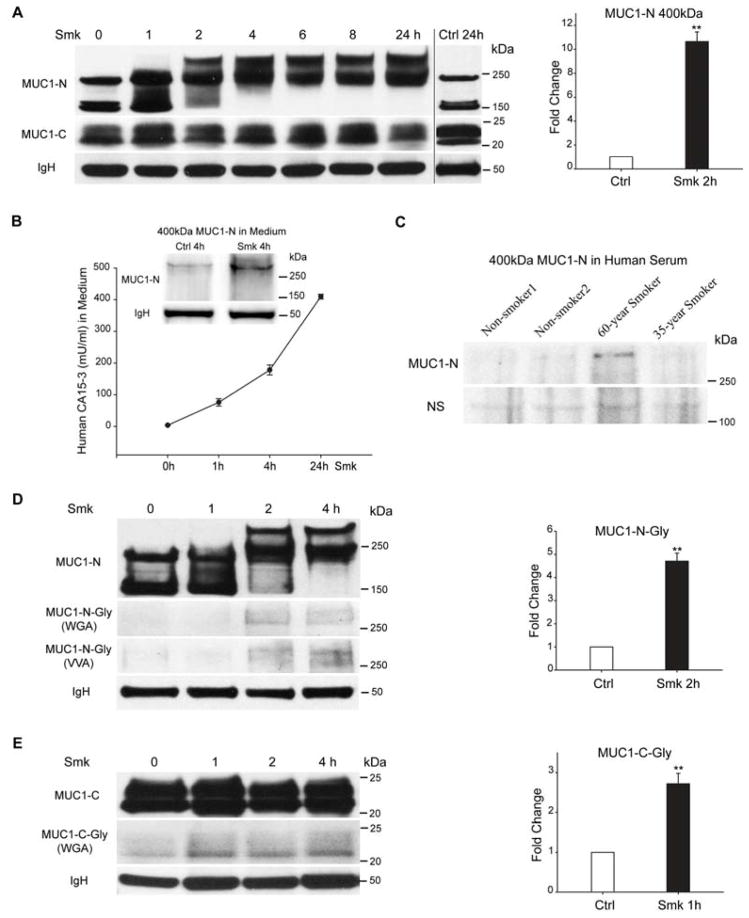
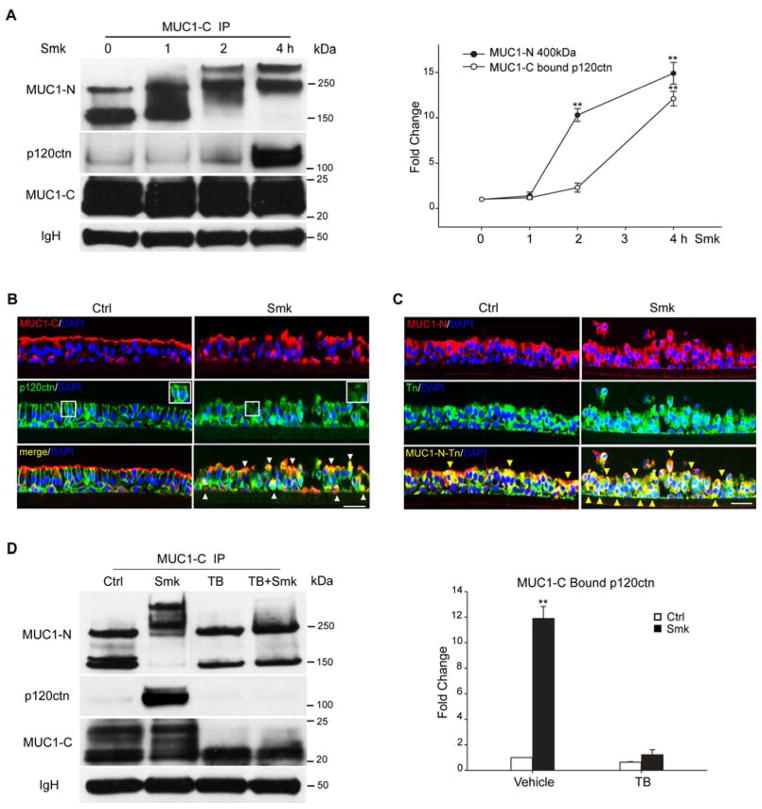
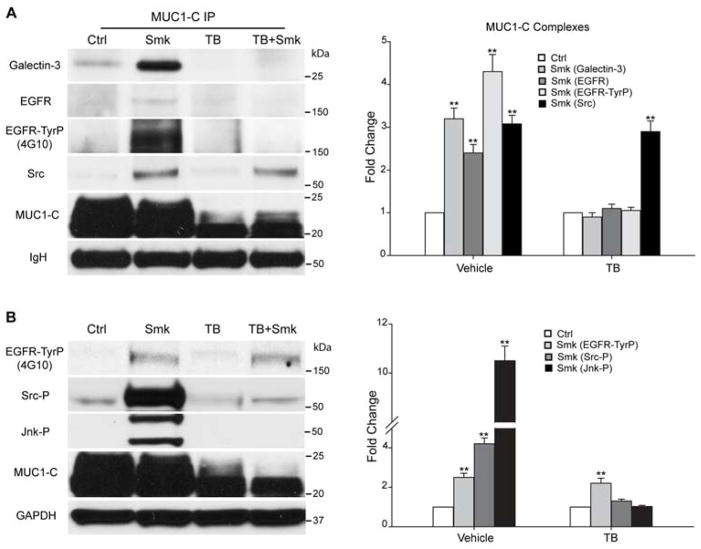
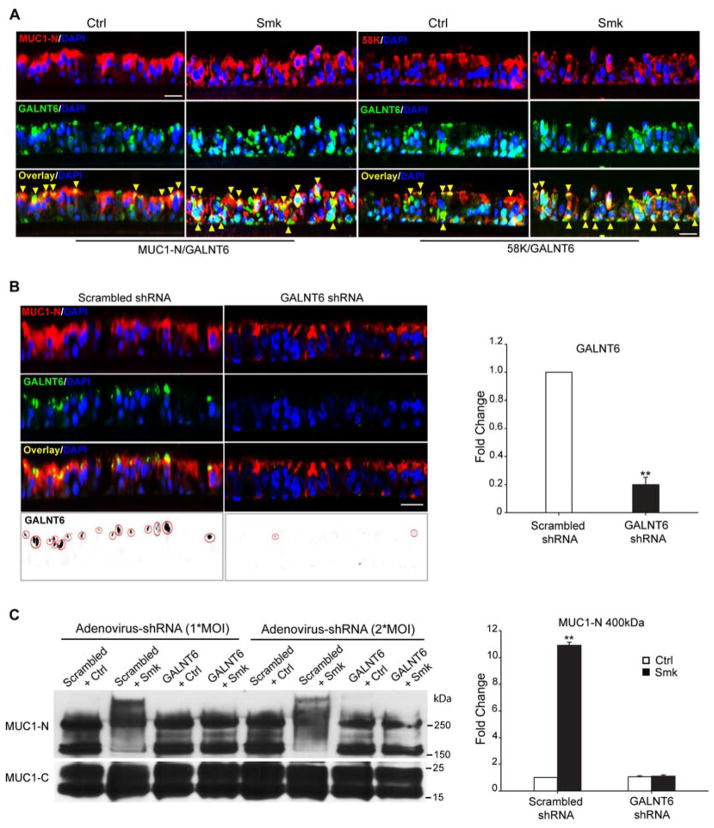
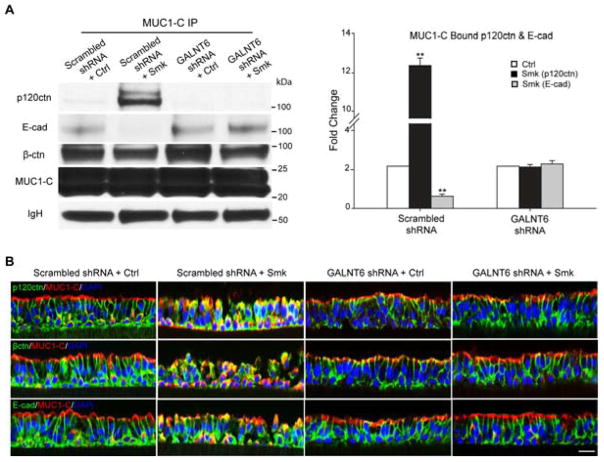
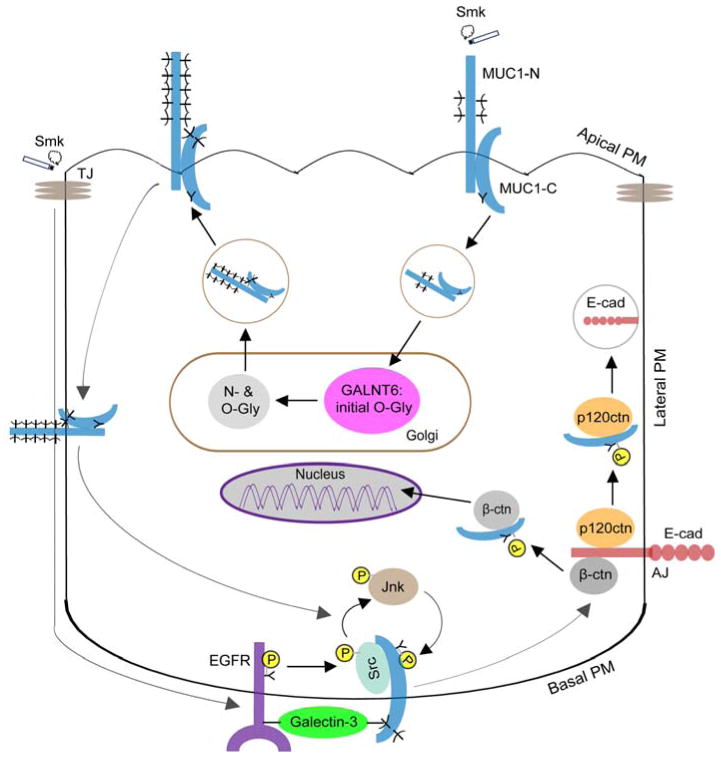
Cigarette smoke increases the risk of lung cancer by 20-fold and accounts for 87% of lung cancer deaths. In the normal airway, heavily O-glycosylated mucin-1 (MUC1) and adherens junctions (AJs) establish a structural barrier that protects the airway from infectious, inflammatory and noxious stimuli. Smoke disrupts cell-cell adhesion via its damaging effects on the AJ protein epithelial cadherin (E-cad). Loss of E-cad is a major hallmark of epithelial-mesenchymal transition (EMT) and has been reported in lung cancer, where it is associated with invasion, metastasis and poor prognosis. Using organotypic cultures of primary human bronchial epithelial (HBE) cells treated with smoke-concentrated medium (Smk), we have demonstrated that E-cad loss is regulated through the aberrant interaction of its AJ binding partner, p120-catenin (p120ctn), and the C-terminus of MUC1 (MUC1-C). Here, we reported that even before MUC1-C became bound to p120ctn, smoke promoted the generation of a novel 400 kDa glycoform of MUC1's N-terminus (MUC1-N) differing from the 230 kDa and 150 kDa glycoforms in untreated control cells. The subsequent smoke-induced, time-dependent shedding of glycosylated MUC1-N exposed MUC1-C as a putative receptor for interactions with EGFR, Src and p120ctn. Smoke-induced MUC1-C glycosylation modulated MUC1-C tyrosine phosphorylation (TyrP) that was essential for MUC1-C/p120ctn interaction through dose-dependent bridging of Src/MUC1-C/galectin-3/EGFR signalosomes. Chemical deglycosylation of MUC1 using a mixture of N-glycosylation inhibitor tunicamycin and O-glycosylation inhibitor benzyl-α-GalNAc disrupted the Src/MUC1-C/galectin-3/EGFR complexes and thereby abolished smoke-induced MUC1-C-TyrP and MUC1-C/p120ctn interaction. Similarly, inhibition of smoke-induced MUC1-N glycosylation using adenoviral shRNA directed against N-acetyl-galactosaminyl transferase-6 (GALNT6, an enzyme that controls the initiating step of O-glycosylation) successfully suppressed MUC1-C/p120ctn interaction, prevented E-cad degradation and maintained cellular polarity in response to smoke. Thus, GALNT6 shRNA represents a potential therapeutic modality to prevent the initiation of events associated with EMT in the smoker's airway.
Keywords: E-cadherin; EGFR; MUC1; cigarette smoke; epithelial-mesenchymal transition; galectin-3; glycosylation; in vitro airway model; lung cancer; p120-catenin.
Copyright © 2014 Pathological Society of Great Britain and Ireland. Published by John Wiley & Sons, Ltd.
Conflict of interest statement
No conflicts of interest were declared.
Figures


Similar articles
-
Cigarette smoke disrupts the integrity of airway adherens junctions through the aberrant interaction of p120-catenin with the cytoplasmic tail of MUC1.J Pathol. 2013 Jan;229(1):74-86. doi: 10.1002/path.4070. Epub 2012 Sep 28. J Pathol. 2013. PMID: 22833523 Free PMC article.
-
Cigarette Smoke Mediates Nuclear to Cytoplasmic Trafficking of Transcriptional Inhibitor Kaiso through MUC1 and P120-Catenin.Am J Pathol. 2016 Dec;186(12):3146-3159. doi: 10.1016/j.ajpath.2016.08.011. Epub 2016 Oct 17. Am J Pathol. 2016. PMID: 27765636 Free PMC article.
-
Cigarette smoke induces epidermal growth factor receptor-dependent redistribution of apical MUC1 and junctional beta-catenin in polarized human airway epithelial cells.Am J Pathol. 2010 Sep;177(3):1255-64. doi: 10.2353/ajpath.2010.091129. Epub 2010 Jul 22. Am J Pathol. 2010. PMID: 20651243 Free PMC article.
-
A central role for cadherin signaling in cancer.Exp Cell Res. 2017 Sep 1;358(1):78-85. doi: 10.1016/j.yexcr.2017.04.006. Epub 2017 Apr 12. Exp Cell Res. 2017. PMID: 28412244 Free PMC article. Review.
-
Functions of p120ctn isoforms in cell-cell adhesion and intracellular signaling.Front Biosci (Landmark Ed). 2012 Jan 1;17(5):1669-94. doi: 10.2741/4012. Front Biosci (Landmark Ed). 2012. PMID: 22201829 Review.
Cited by
-
Extracellular galectin-3 programs multidrug resistance through Na+/K+-ATPase and P-glycoprotein signaling.Oncotarget. 2015 Aug 14;6(23):19592-604. doi: 10.18632/oncotarget.4285. Oncotarget. 2015. PMID: 26158764 Free PMC article.
-
Mucin Glycans: A Target for Cancer Therapy.Molecules. 2023 Oct 11;28(20):7033. doi: 10.3390/molecules28207033. Molecules. 2023. PMID: 37894512 Free PMC article. Review.
-
Murine Trophoblast Stem Cells and Their Differentiated Cells Attenuate Zika Virus In Vitro by Reducing Glycosylation of the Viral Envelope Protein.Cells. 2021 Nov 9;10(11):3085. doi: 10.3390/cells10113085. Cells. 2021. PMID: 34831310 Free PMC article.
-
Systematic analysis of the human tumor cell binding to human vs. murine E- and P-selectin under static vs. dynamic conditions.Glycobiology. 2020 Aug 20;30(9):695-709. doi: 10.1093/glycob/cwaa019. Glycobiology. 2020. PMID: 32103235 Free PMC article.
-
Polypeptide N-acetylgalactosaminyltransferase-6 expression independently predicts poor overall survival in patients with lung adenocarcinoma after curative resection.Oncotarget. 2016 Aug 23;7(34):54463-54473. doi: 10.18632/oncotarget.9810. Oncotarget. 2016. PMID: 27276675 Free PMC article.
References
-
- Society AC. Cancer Facts & Figures 2009. 2009 Internet http://wwwcancerorg/acs/groups/content/@nho/documents/document/500809web....
-
- Gilpin EA, Pierce JP. Demographic differences in patterns in the incidence of smoking cessation: United States 1950–1990. Ann Epidemiol. 2002;12:141–150. - PubMed
-
- Polyak K, Weinberg RA. Transitions between epithelial and mesenchymal states: acquisition of malignant and stem cell traits. Nat Rev Cancer. 2009;9:265–273. - PubMed
-
- Thoreson MA, Reynolds AB. Altered expression of the catenin p120 in human cancer: implications for tumor progression. Differentiation. 2002;70:583–589. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous
