

Inferring fitness landscapes by regression produces biased estimates of epistasis
- PMID: 24843135
- PMCID: PMC4050575
- DOI: 10.1073/pnas.1400849111
Inferring fitness landscapes by regression produces biased estimates of epistasis
Abstract
The genotype-fitness map plays a fundamental role in shaping the dynamics of evolution. However, it is difficult to directly measure a fitness landscape in practice, because the number of possible genotypes is astronomical. One approach is to sample as many genotypes as possible, measure their fitnesses, and fit a statistical model of the landscape that includes additive and pairwise interactive effects between loci. Here, we elucidate the pitfalls of using such regressions by studying artificial but mathematically convenient fitness landscapes. We identify two sources of bias inherent in these regression procedures, each of which tends to underestimate high fitnesses and overestimate low fitnesses. We characterize these biases for random sampling of genotypes as well as samples drawn from a population under selection in the Wright-Fisher model of evolutionary dynamics. We show that common measures of epistasis, such as the number of monotonically increasing paths between ancestral and derived genotypes, the prevalence of sign epistasis, and the number of local fitness maxima, are distorted in the inferred landscape. As a result, the inferred landscape will provide systematically biased predictions for the dynamics of adaptation. We identify the same biases in a computational RNA-folding landscape as well as regulatory sequence binding data treated with the same fitting procedure. Finally, we present a method to ameliorate these biases in some cases.
Keywords: experimental evolution; molecular evolution; penalized regression.
Conflict of interest statement
The authors declare no conflict of interest.
Figures






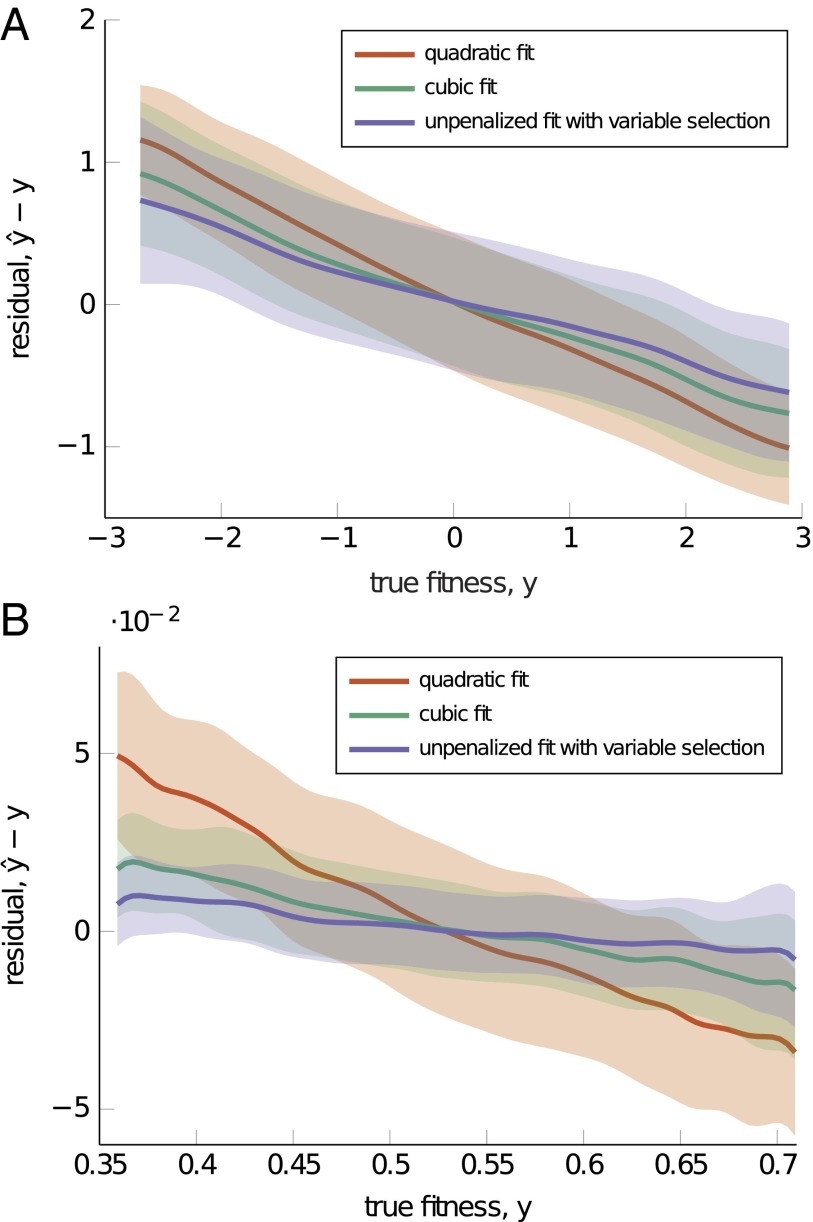
References
-
- Lenski RE, Rose MR, Simpson SC, Tadler SC. Long-term experimental evolution in Escherichia coli. I. Adaptation and divergence during 2,000 generations. Am Nat. 1991;138(6):1315.
-
- Elena SF, Lenski RE. Evolution experiments with microorganisms: The dynamics and genetic bases of adaptation. Nat Rev Genet. 2003;4(6):457–469. - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
