

Relating the metatranscriptome and metagenome of the human gut
- PMID: 24843156
- PMCID: PMC4050606
- DOI: 10.1073/pnas.1319284111
Relating the metatranscriptome and metagenome of the human gut
Abstract
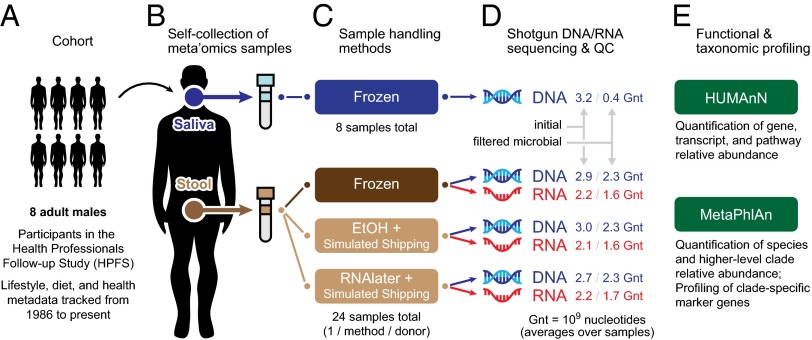
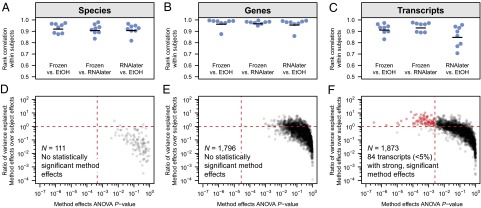
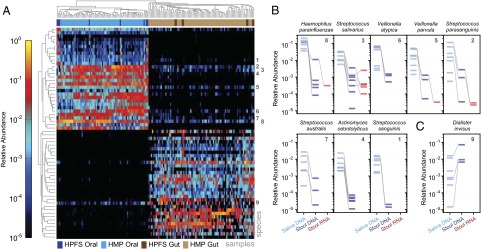
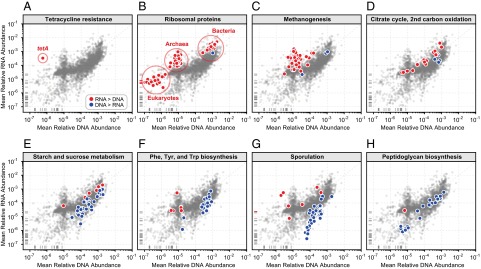
Although the composition of the human microbiome is now well-studied, the microbiota's >8 million genes and their regulation remain largely uncharacterized. This knowledge gap is in part because of the difficulty of acquiring large numbers of samples amenable to functional studies of the microbiota. We conducted what is, to our knowledge, one of the first human microbiome studies in a well-phenotyped prospective cohort incorporating taxonomic, metagenomic, and metatranscriptomic profiling at multiple body sites using self-collected samples. Stool and saliva were provided by eight healthy subjects, with the former preserved by three different methods (freezing, ethanol, and RNAlater) to validate self-collection. Within-subject microbial species, gene, and transcript abundances were highly concordant across sampling methods, with only a small fraction of transcripts (<5%) displaying between-method variation. Next, we investigated relationships between the oral and gut microbial communities, identifying a subset of abundant oral microbes that routinely survive transit to the gut, but with minimal transcriptional activity there. Finally, systematic comparison of the gut metagenome and metatranscriptome revealed that a substantial fraction (41%) of microbial transcripts were not differentially regulated relative to their genomic abundances. Of the remainder, consistently underexpressed pathways included sporulation and amino acid biosynthesis, whereas up-regulated pathways included ribosome biogenesis and methanogenesis. Across subjects, metatranscriptional profiles were significantly more individualized than DNA-level functional profiles, but less variable than microbial composition, indicative of subject-specific whole-community regulation. The results thus detail relationships between community genomic potential and gene expression in the gut, and establish the feasibility of metatranscriptomic investigations in subject-collected and shipped samples.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
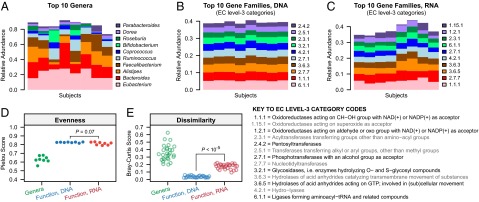
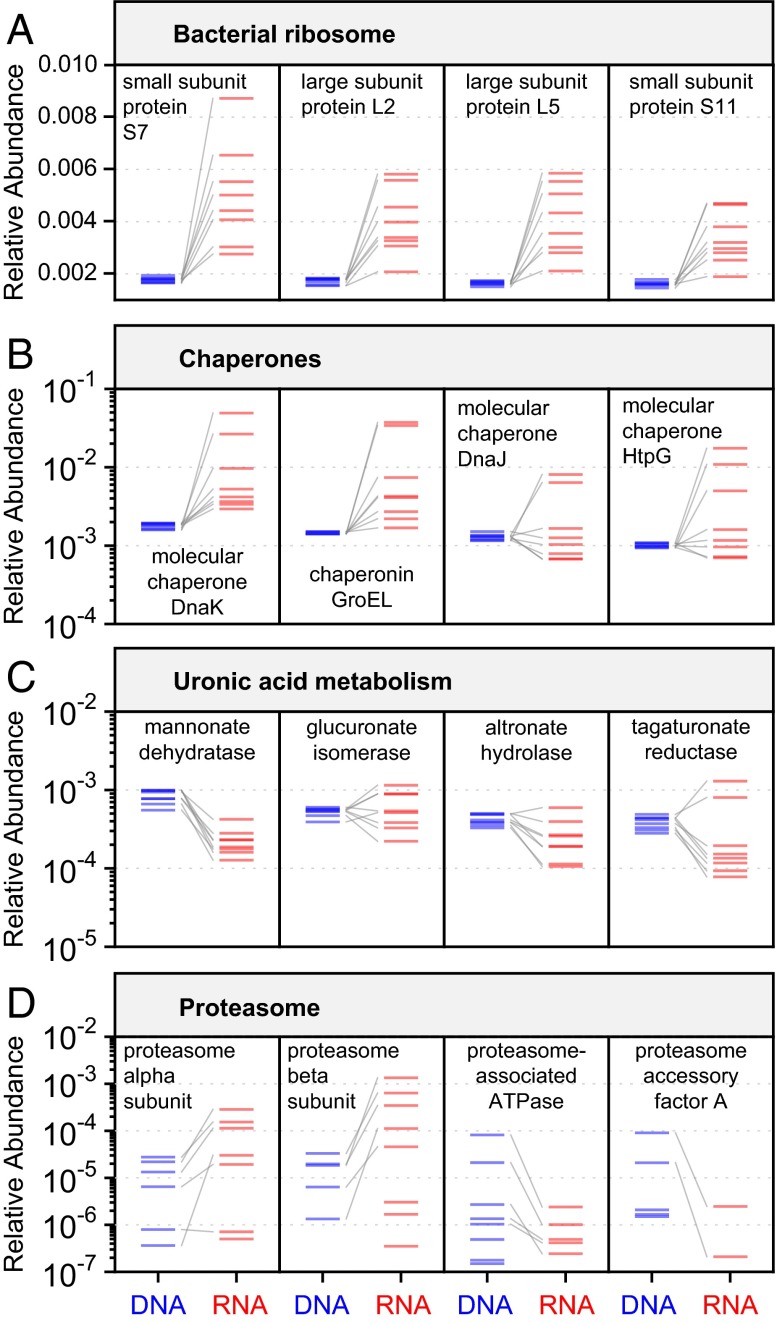
References
-
- Shi Y, Tyson GW, DeLong EF. Metatranscriptomics reveals unique microbial small RNAs in the ocean’s water column. Nature. 2009;459(7244):266–269. - PubMed
Publication types
MeSH terms
Substances
Associated data
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
