

Epigenetic dysregulation in glioma
- PMID: 24843883
- PMCID: PMC4317798
- DOI: 10.1111/cas.12379
Epigenetic dysregulation in glioma
Abstract
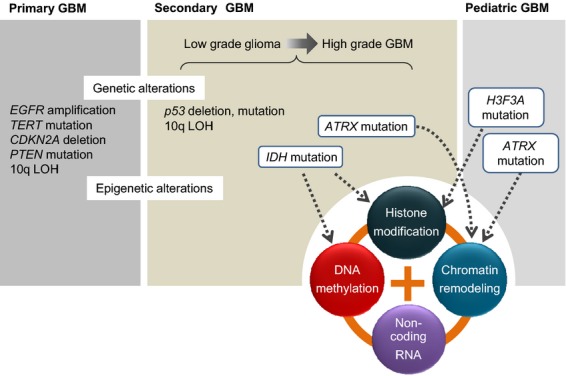
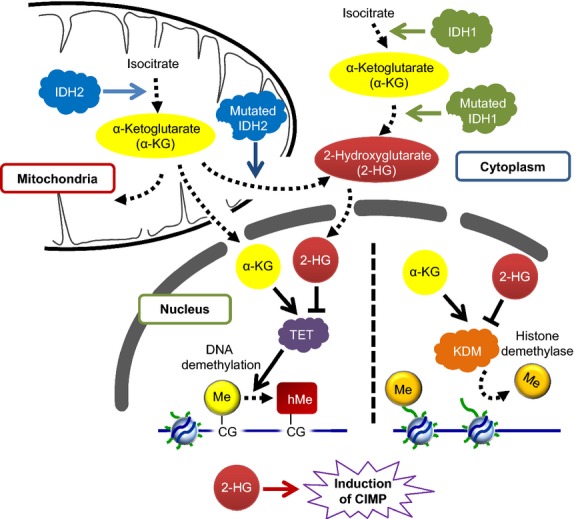
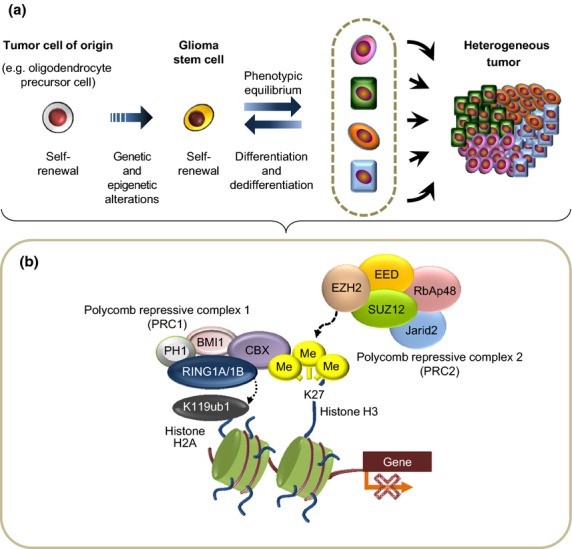
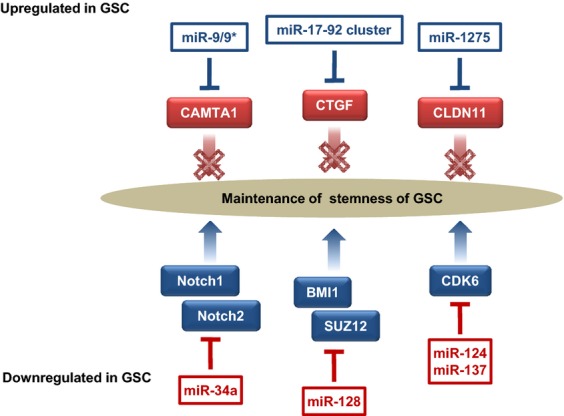
Given that treatment options for patients with glioblastoma are limited, much effort has been made to clarify the underlying mechanisms of gliomagenesis. Recent genome-wide genomic and epigenomic analyses have revealed that mutations in epigenetic modifiers occur frequently in gliomas and that dysregulation of epigenetic mechanisms is closely associated with glioma formation. Given that epigenetic changes are reversible, understanding the epigenetic abnormalities that arise in gliomagenesis might be key to developing more effective treatment strategies for glioma. In this review, we focus on the recent advancements in epigenetic research with respect to gliomas, consider how epigenetic mechanisms dynamically regulate tumor cells, including the cancer stem cell population, and discuss perspectives and challenges for glioma treatment in the near future.
Figures
References
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
