

Anti-diabetic activity of insulin-degrading enzyme inhibitors mediated by multiple hormones
- PMID: 24847884
- PMCID: PMC4142213
- DOI: 10.1038/nature13297
Anti-diabetic activity of insulin-degrading enzyme inhibitors mediated by multiple hormones
Abstract
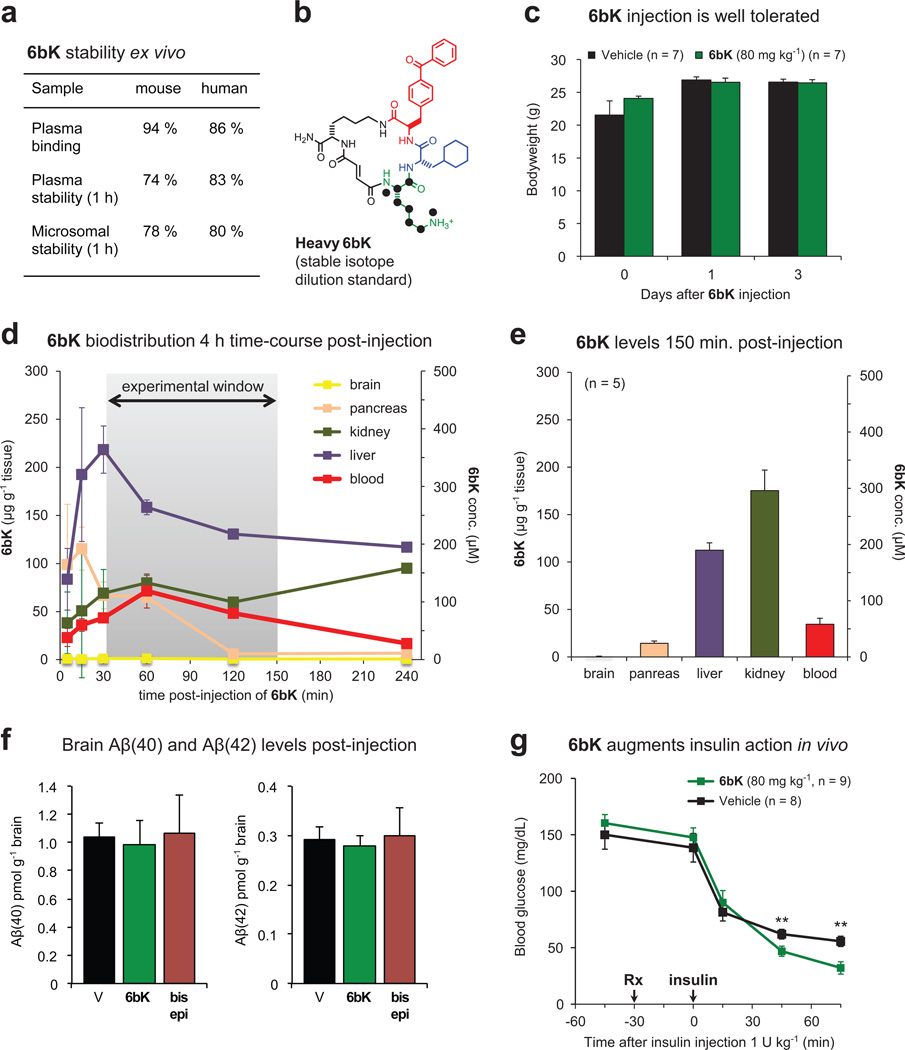
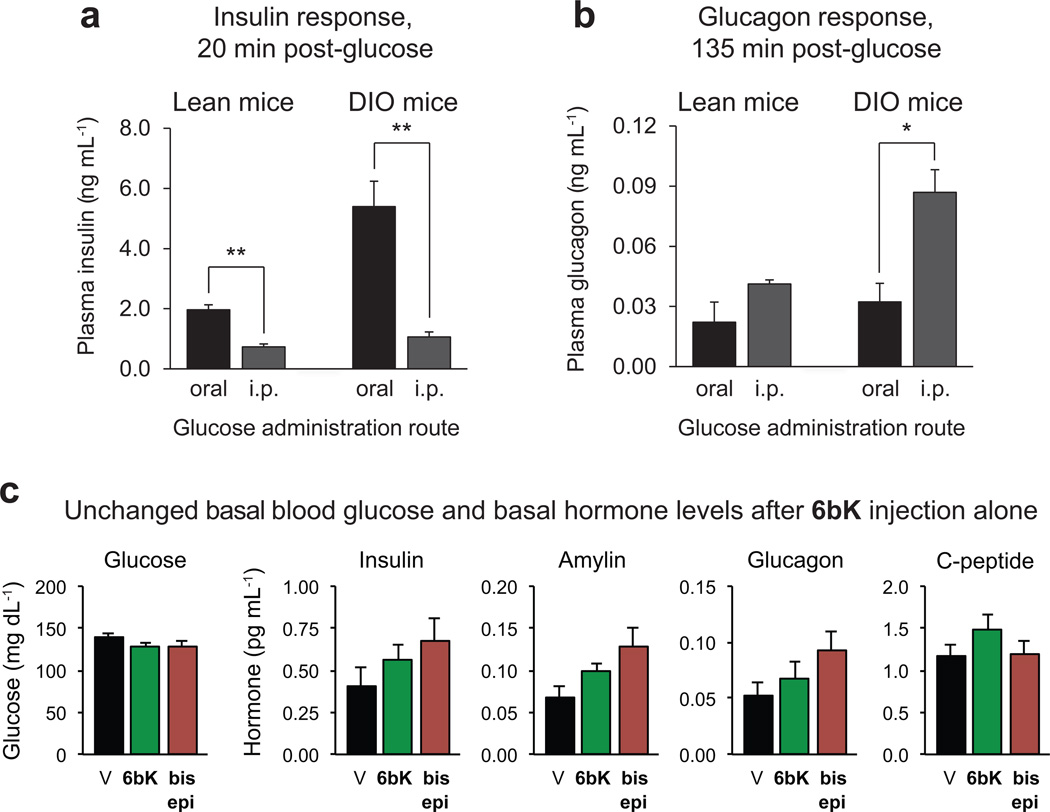
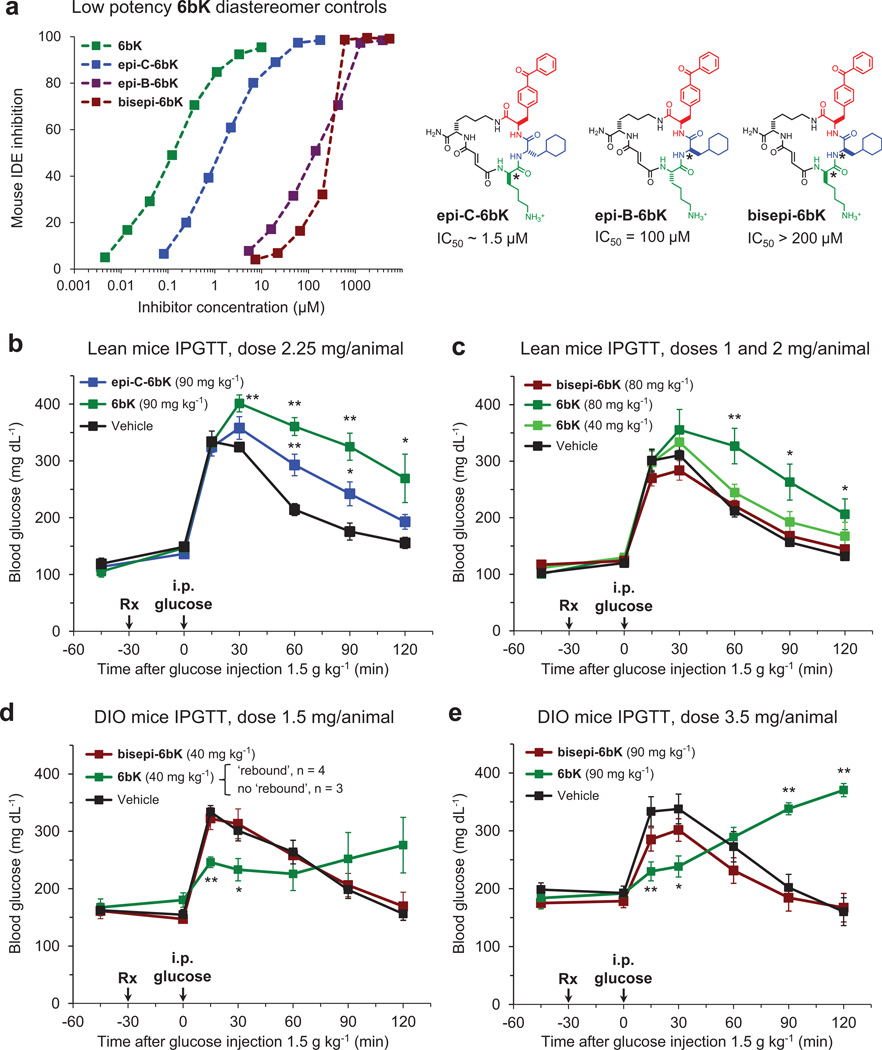
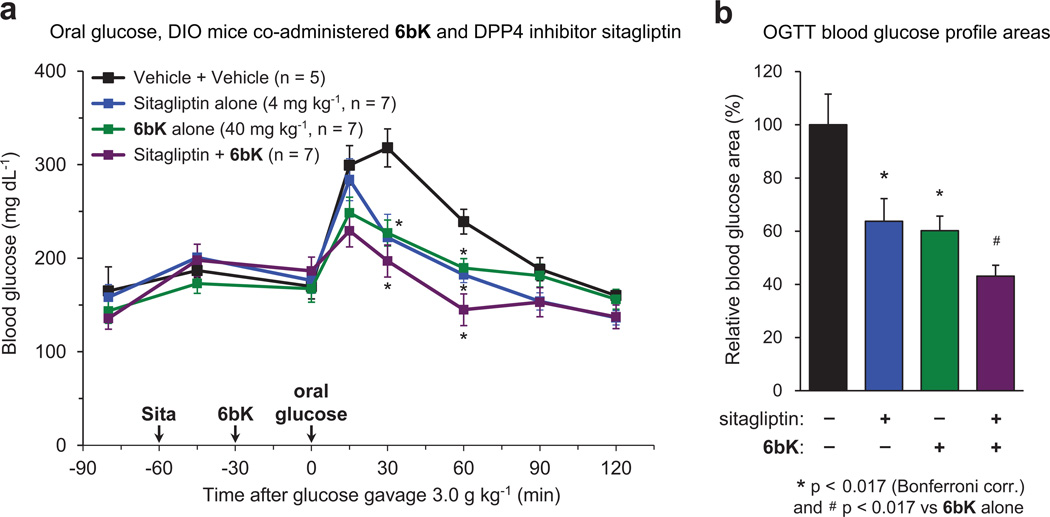
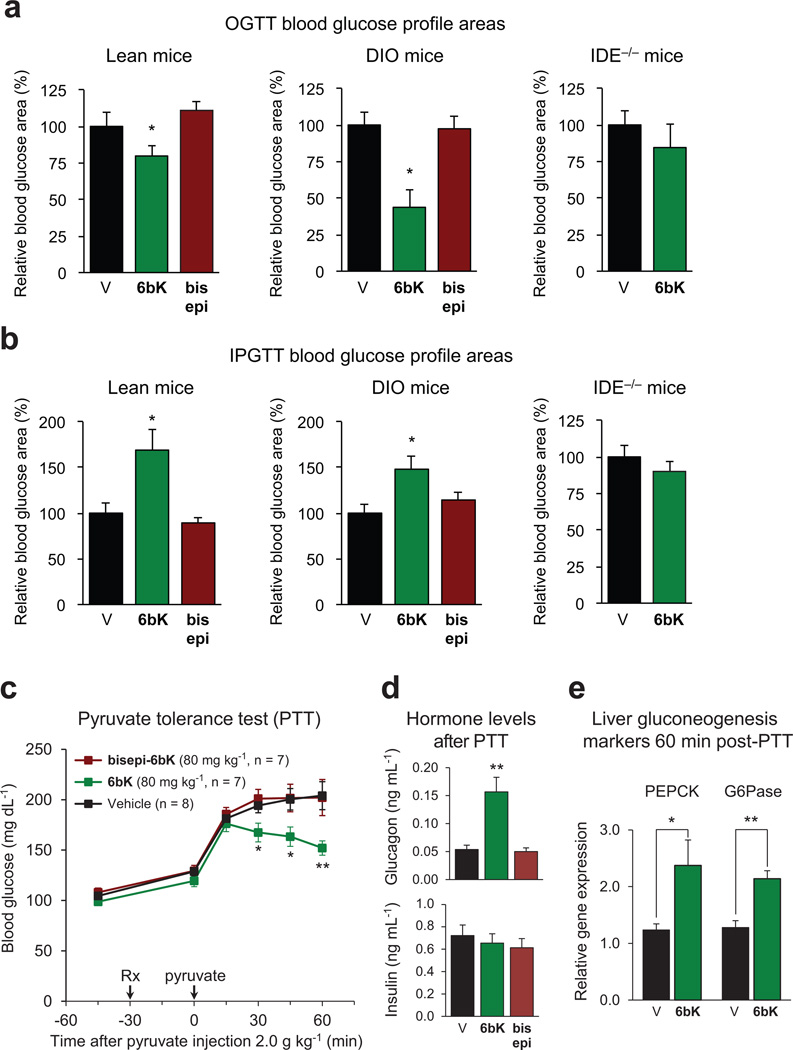
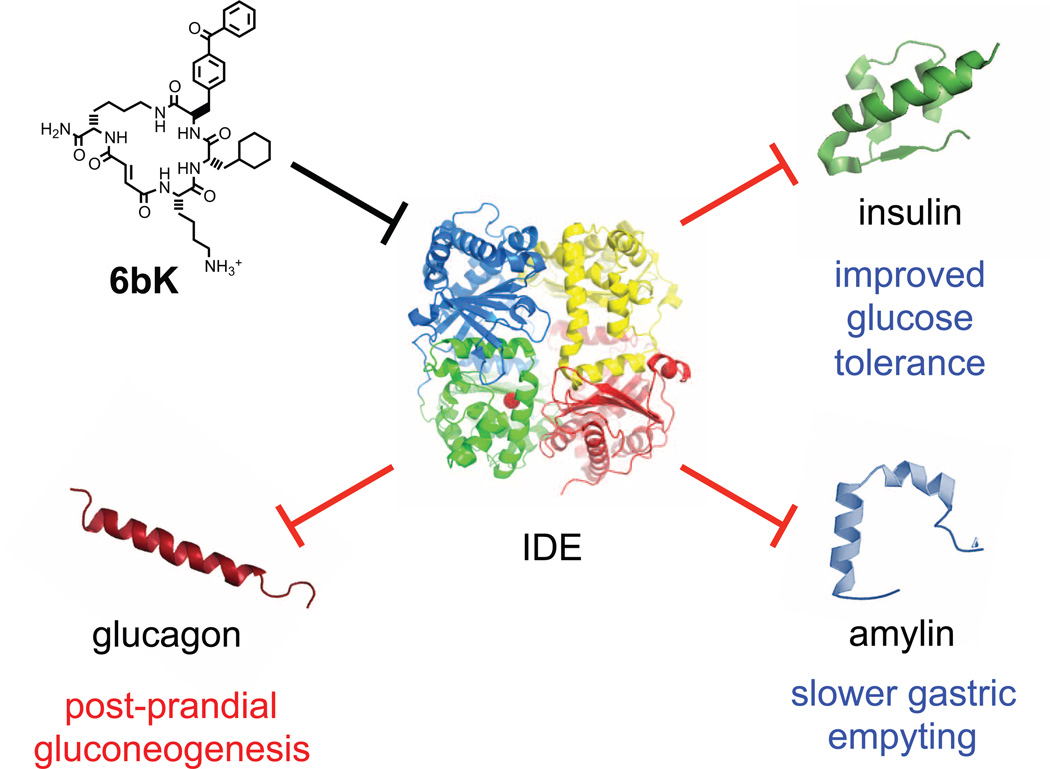
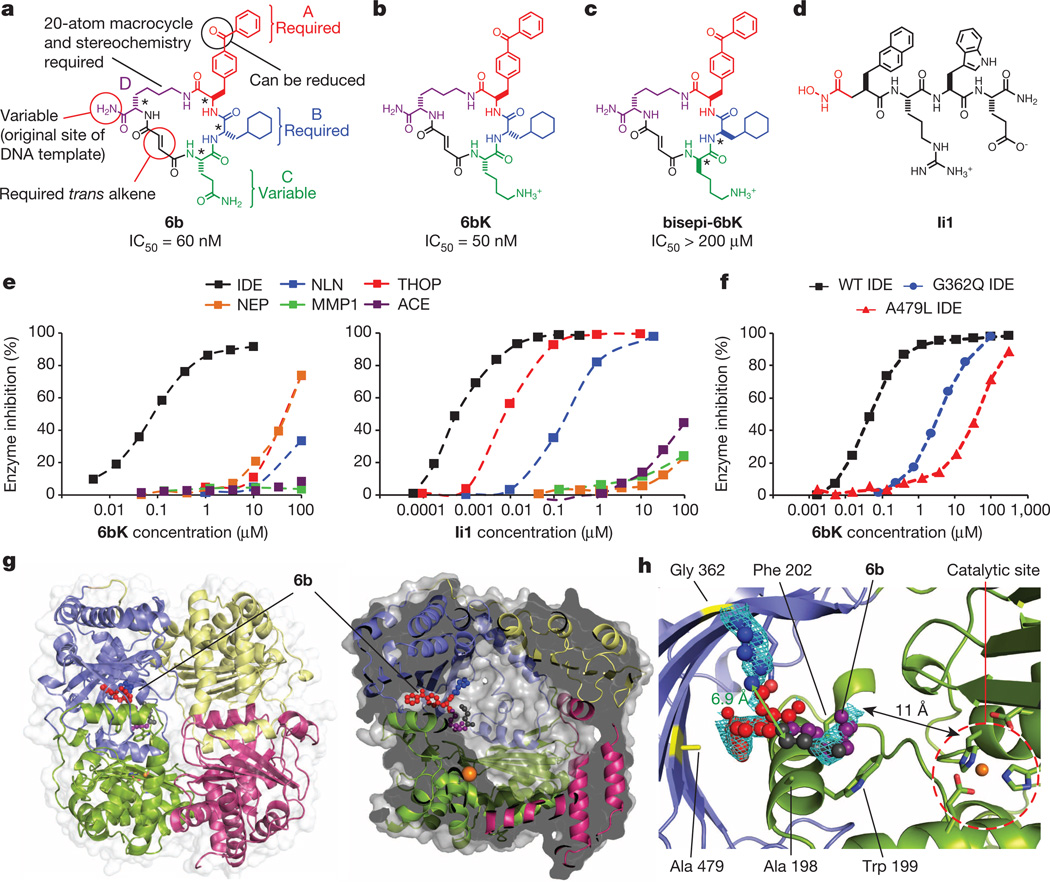
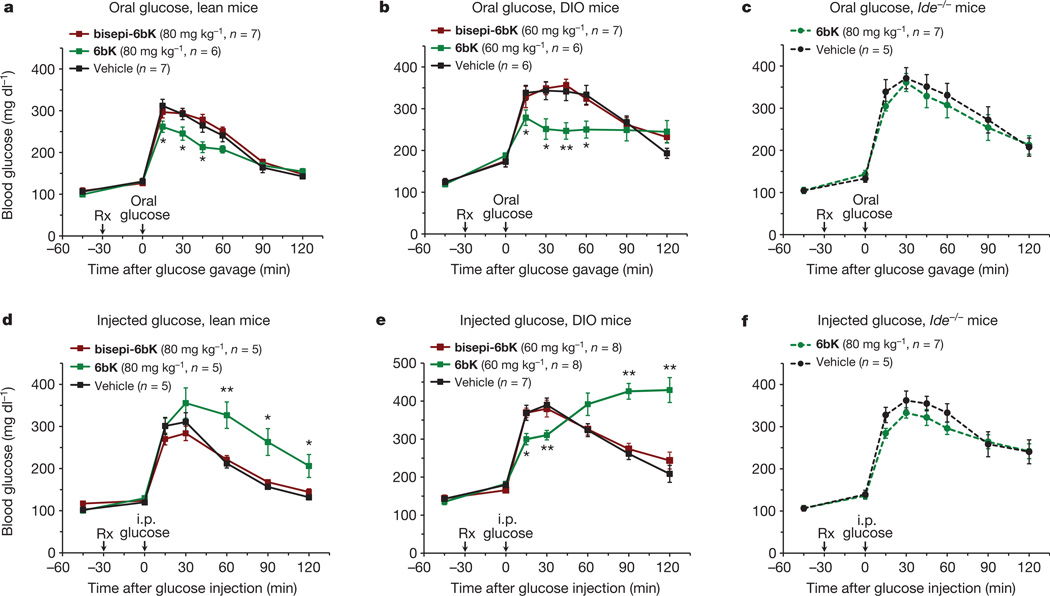
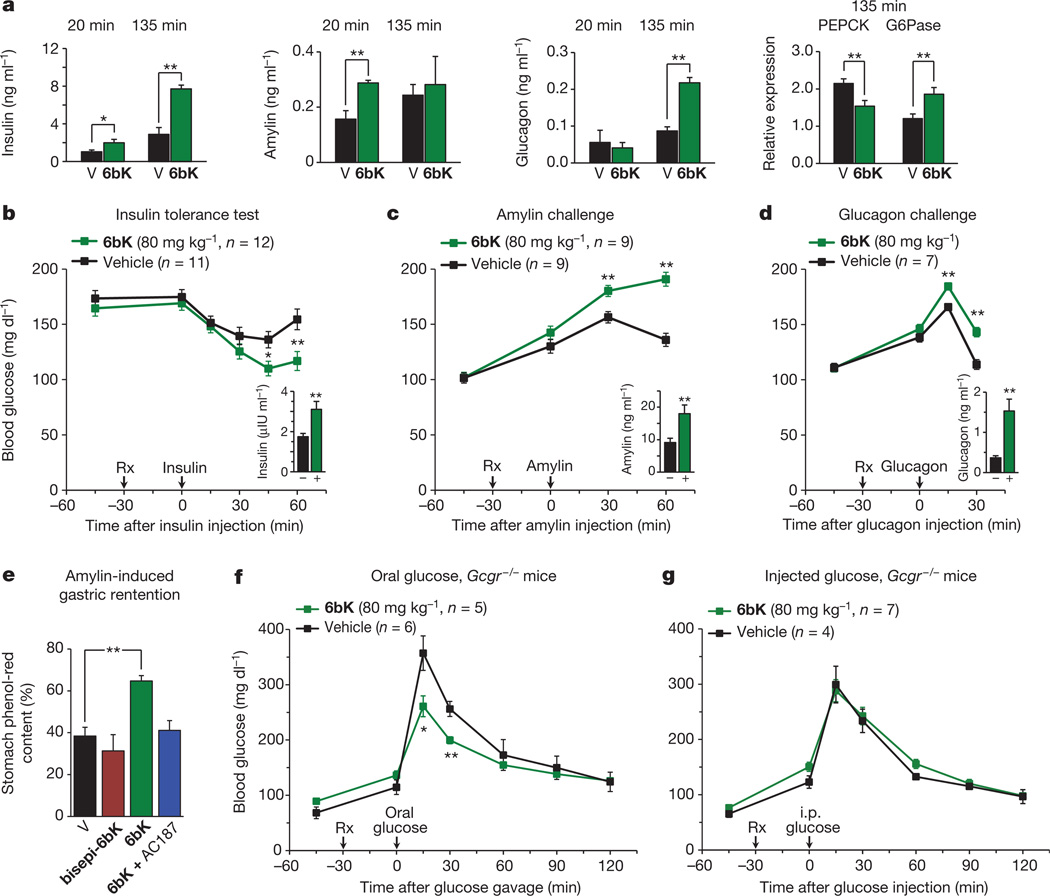
Despite decades of speculation that inhibiting endogenous insulin degradation might treat type-2 diabetes, and the identification of IDE (insulin-degrading enzyme) as a diabetes susceptibility gene, the relationship between the activity of the zinc metalloprotein IDE and glucose homeostasis remains unclear. Although Ide(-/-) mice have elevated insulin levels, they exhibit impaired, rather than improved, glucose tolerance that may arise from compensatory insulin signalling dysfunction. IDE inhibitors that are active in vivo are therefore needed to elucidate IDE's physiological roles and to determine its potential to serve as a target for the treatment of diabetes. Here we report the discovery of a physiologically active IDE inhibitor identified from a DNA-templated macrocycle library. An X-ray structure of the macrocycle bound to IDE reveals that it engages a binding pocket away from the catalytic site, which explains its remarkable selectivity. Treatment of lean and obese mice with this inhibitor shows that IDE regulates the abundance and signalling of glucagon and amylin, in addition to that of insulin. Under physiological conditions that augment insulin and amylin levels, such as oral glucose administration, acute IDE inhibition leads to substantially improved glucose tolerance and slower gastric emptying. These findings demonstrate the feasibility of modulating IDE activity as a new therapeutic strategy to treat type-2 diabetes and expand our understanding of the roles of IDE in glucose and hormone regulation.
Figures
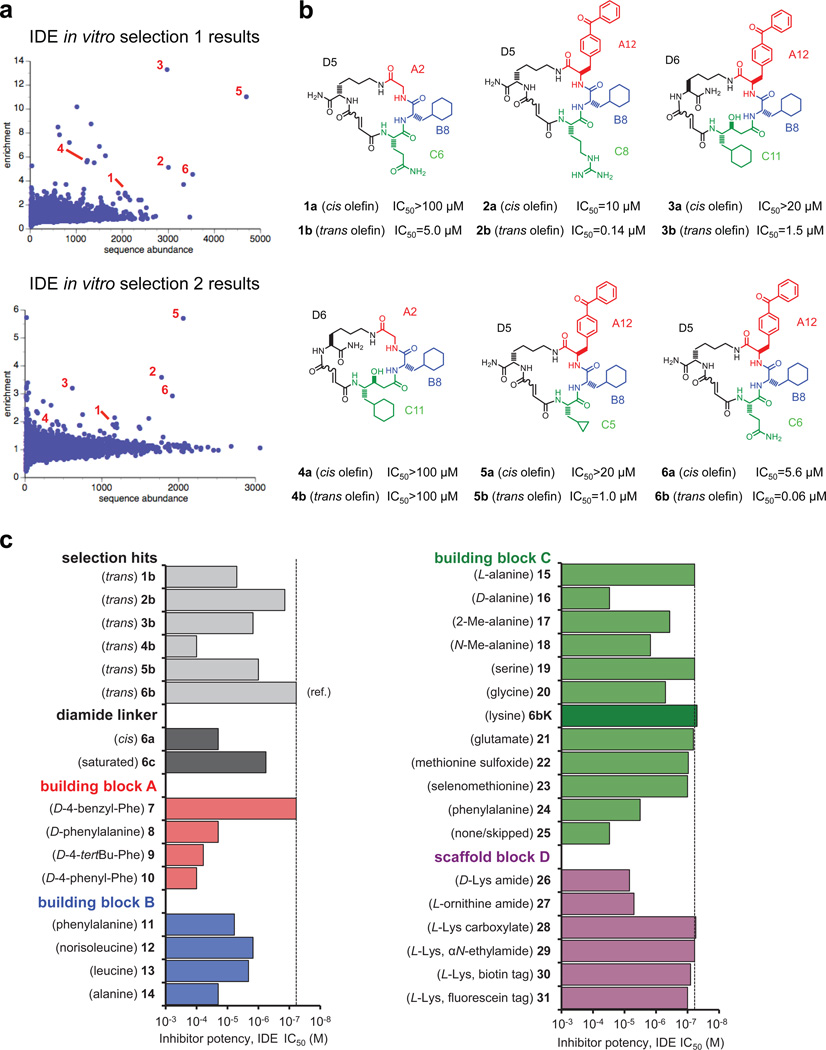
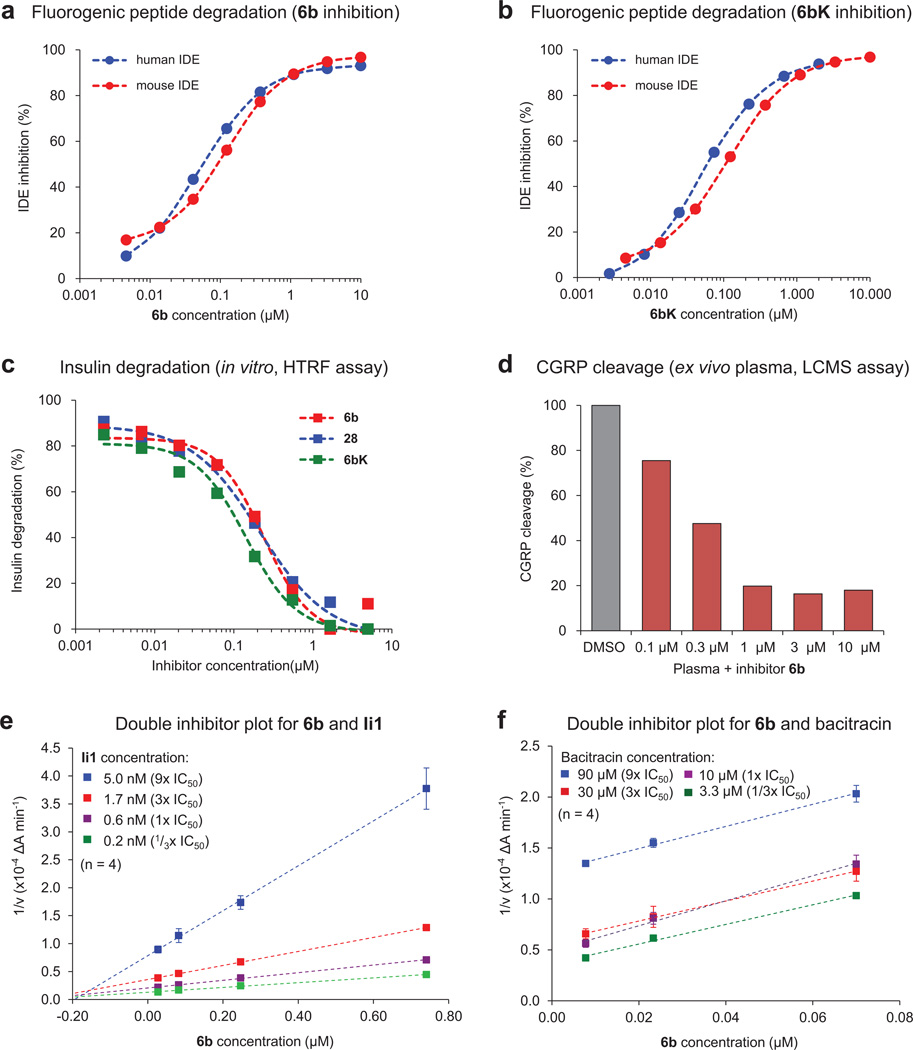
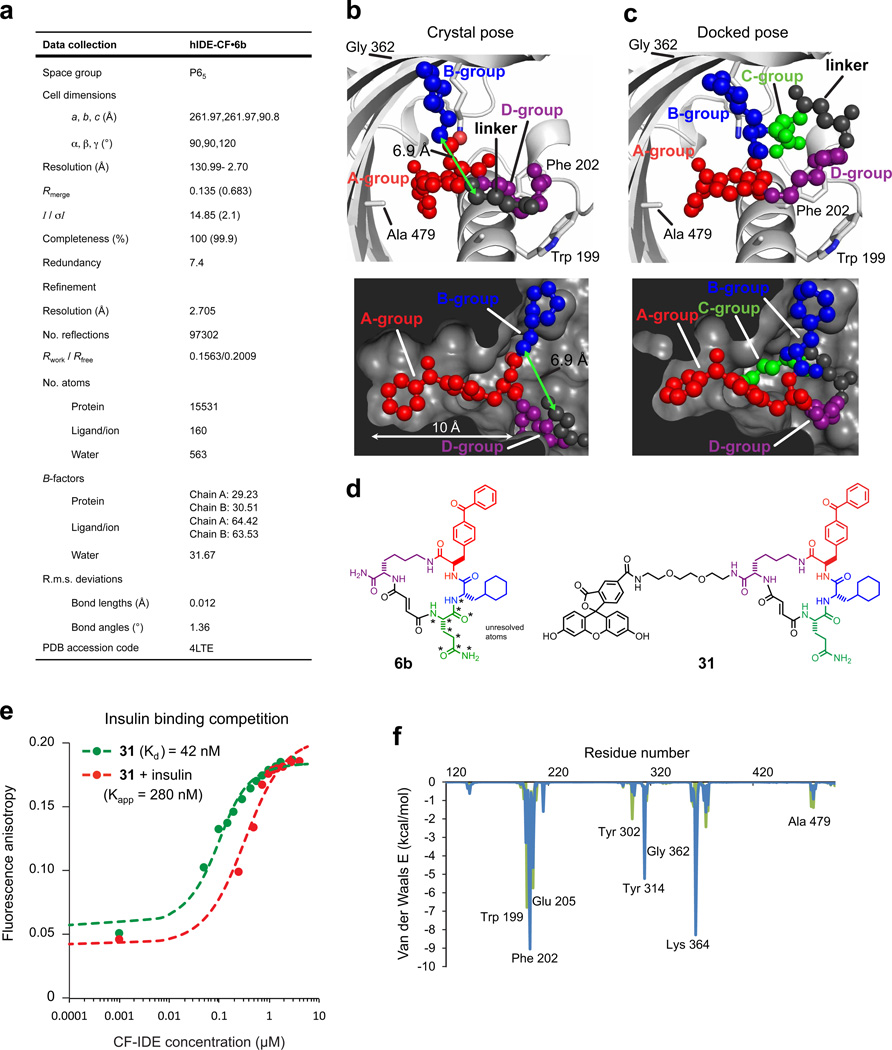
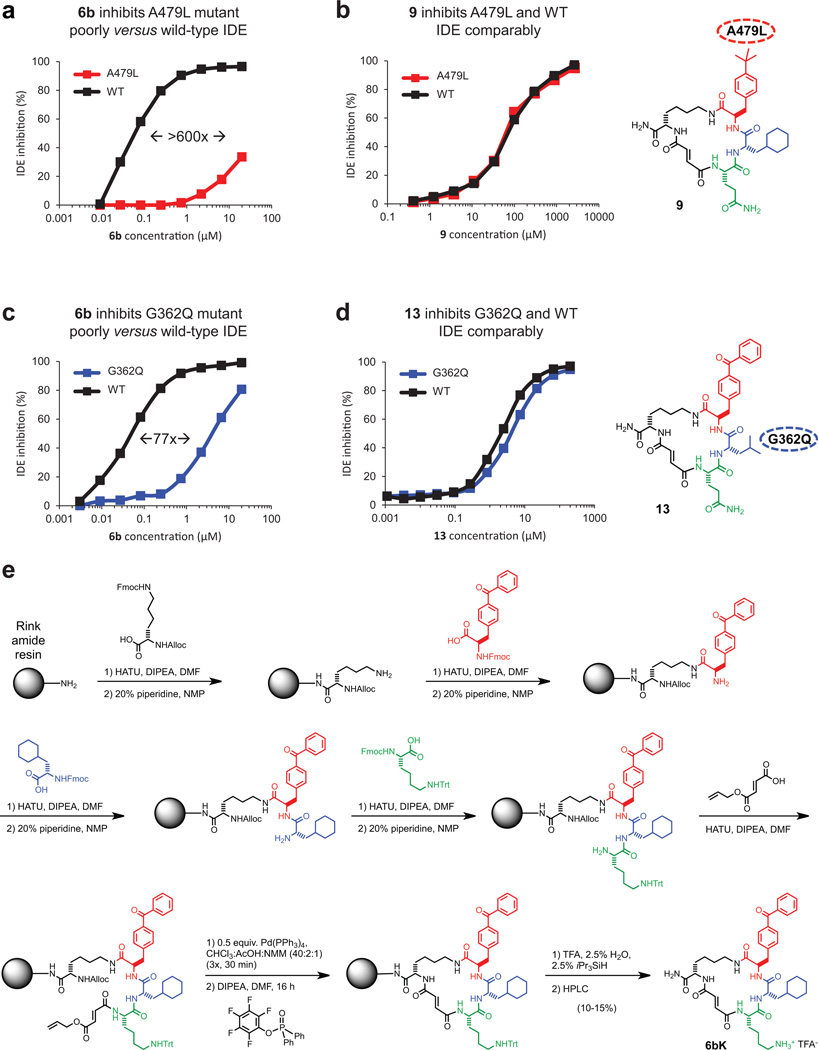
Comment in
-
Insulin-degrading enzyme inhibition, a novel therapy for type 2 diabetes?Cell Metab. 2014 Aug 5;20(2):201-3. doi: 10.1016/j.cmet.2014.07.016. Cell Metab. 2014. PMID: 25100059 Free PMC article.
References
-
- Duckworth WC, Bennett RG, Hamel FG. Insulin degradation: progress and potential. Endocr. Rev. 1998;19:608–624. - PubMed
-
- Mirsky IA, Broh-Kahn RH. The inactivation of insulin by tissue extracts the distribution and properties of insulin inactivating extracts. Arch. Biochem. 1949;20:1–9. - PubMed
-
- Sladek R, et al. A genome-wide association study identifies novel risk loci for type 2 diabetes. Nature. 2007;445:881–885. - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
Grants and funding
- UL1 TR000430/TR/NCATS NIH HHS/United States
- R01 GM081539/GM/NIGMS NIH HHS/United States
- T32 GM008444/GM/NIGMS NIH HHS/United States
- T32 GM007598/GM/NIGMS NIH HHS/United States
- R00 GM080097/GM/NIGMS NIH HHS/United States
- DP2 OD002374/OD/NIH HHS/United States
- HHMI/Howard Hughes Medical Institute/United States
- P41 GM111244/GM/NIGMS NIH HHS/United States
- P30 DK057521/DK/NIDDK NIH HHS/United States
- F30 CA174152/CA/NCI NIH HHS/United States
- R01 GM81539/GM/NIGMS NIH HHS/United States
- R01 GM065865/GM/NIGMS NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
