

A single amino acid in EBNA-2 determines superior B lymphoblastoid cell line growth maintenance by Epstein-Barr virus type 1 EBNA-2
- PMID: 24850736
- PMCID: PMC4136291
- DOI: 10.1128/JVI.01000-14
A single amino acid in EBNA-2 determines superior B lymphoblastoid cell line growth maintenance by Epstein-Barr virus type 1 EBNA-2
Abstract
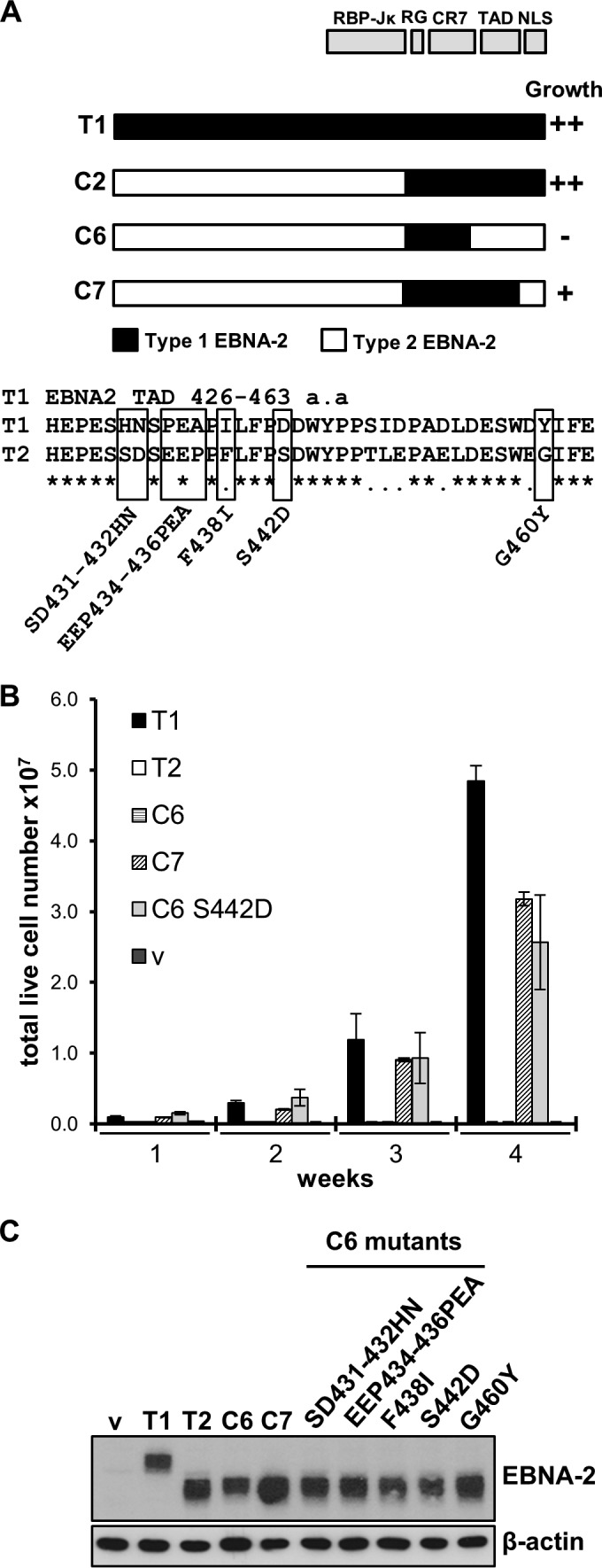
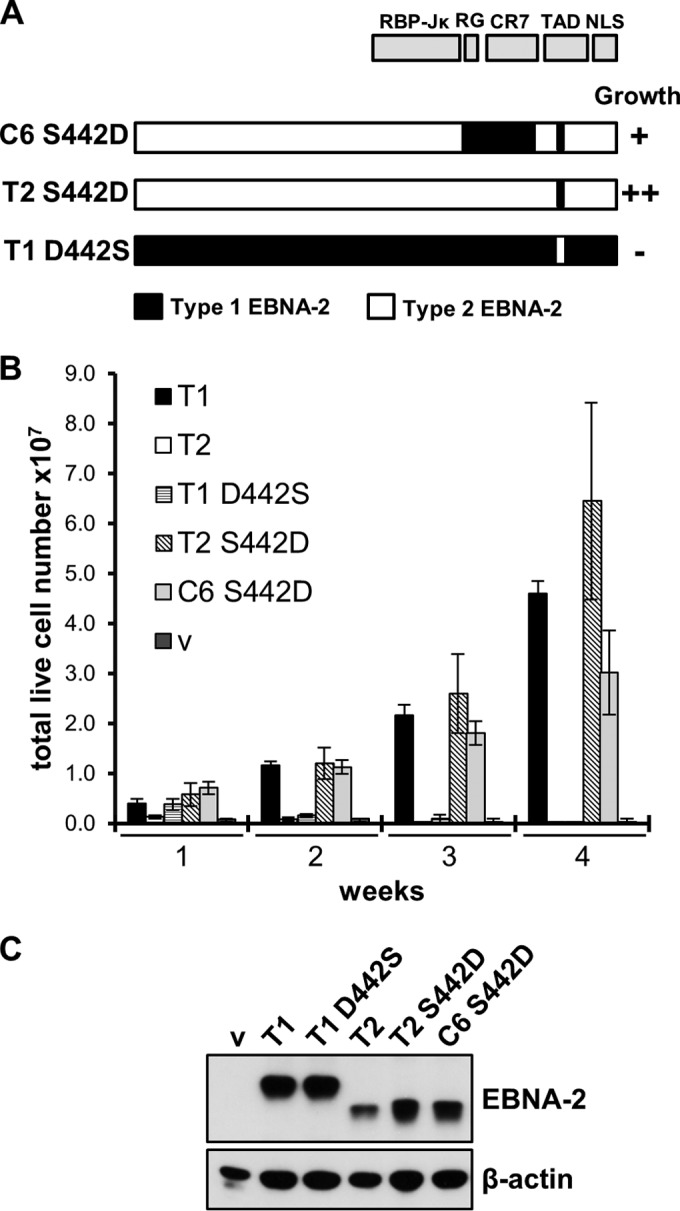
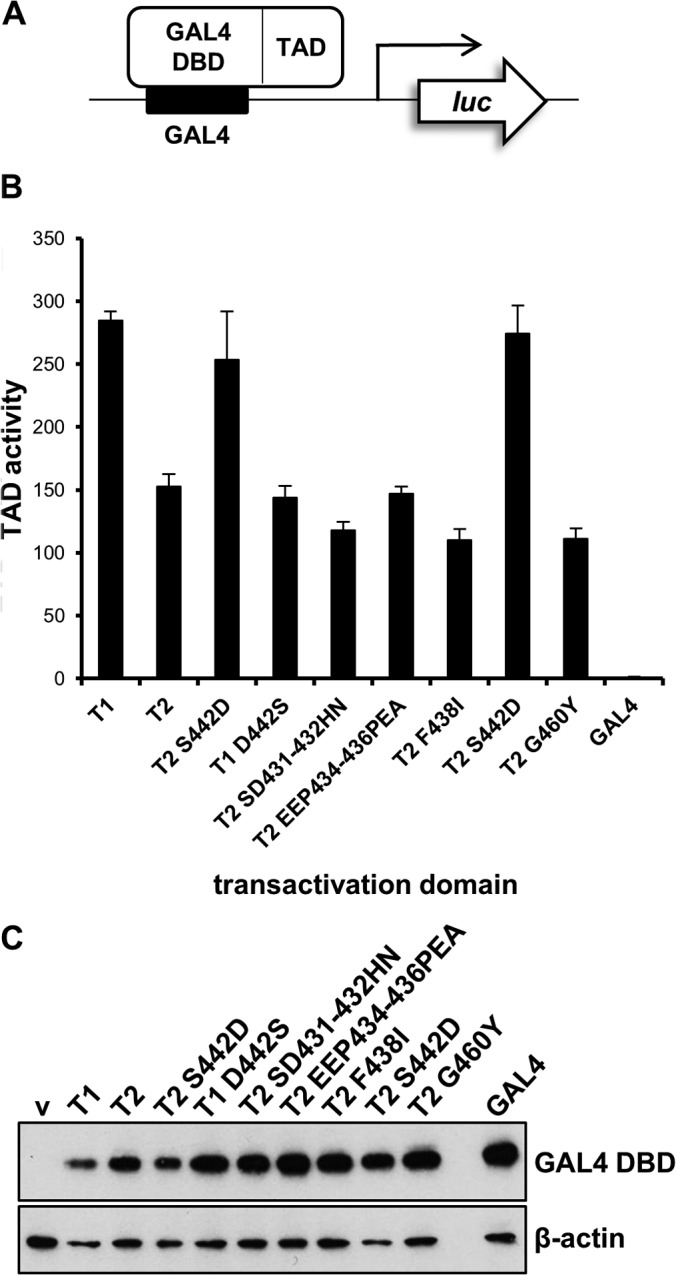
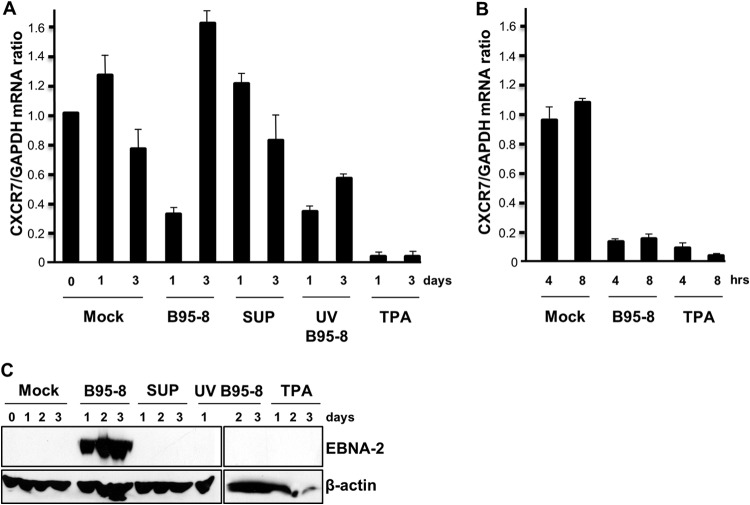
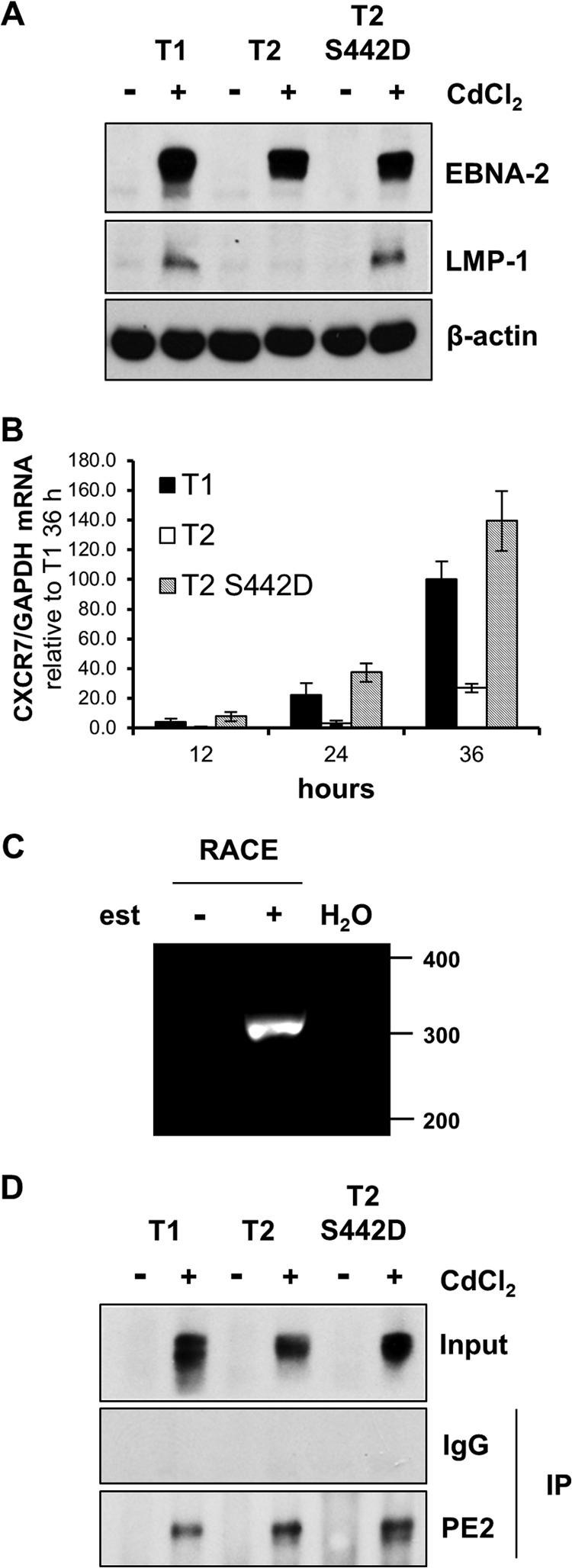
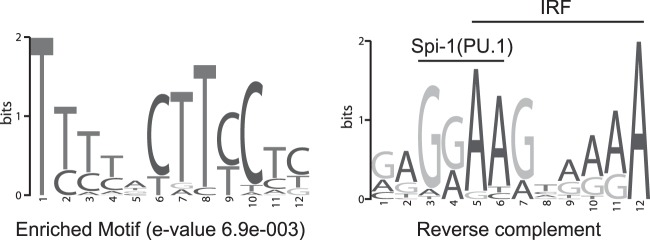
Sequence differences in the EBNA-2 protein mediate the superior ability of type 1 Epstein-Barr virus (EBV) to transform human B cells into lymphoblastoid cell lines compared to that of type 2 EBV. Here we show that changing a single amino acid (S442D) from serine in type 2 EBNA-2 to the aspartate found in type 1 EBNA-2 confers a type 1 growth phenotype in a lymphoblastoid cell line growth maintenance assay. This amino acid lies in the transactivation domain of EBNA-2, and the S442D change increases activity in a transactivation domain assay. The superior growth properties of type 1 EBNA-2 correlate with the greater induction of EBV LMP-1 and about 10 cell genes, including CXCR7. In chromatin immunoprecipitation assays, type 1 EBNA-2 is shown to associate more strongly with EBNA-2 binding sites near the LMP-1 and CXCR7 genes. Unbiased motif searching of the EBNA-2 binding regions of the differentially regulated cell genes identified an ETS-interferon regulatory factor composite element motif that closely corresponds to the sequences known to mediate EBNA-2 regulation of the LMP-1 promoter. It appears that the superior induction by type 1 EBNA-2 of the cell genes contributing to cell growth is due to their being regulated in a manner different from that for most EBNA-2-responsive genes and in a way similar to that for the LMP-1 gene.
Importance: The EBNA-2 transcription factor plays a key role in B cell transformation by EBV and defines the two EBV types. Here we identify a single amino acid (Ser in type 1 EBV, Asp in type 2 EBV) of EBNA-2 that determines the superior ability of type 1 EBNA-2 to induce a key group of cell genes and the EBV LMP-1 gene, which mediate the growth advantage of B cells infected with type 1 EBV. The EBNA-2 binding sites in these cell genes have a sequence motif similar to the sequence known to mediate regulation of the EBV LMP-1 promoter. Further detailed analysis of transactivation and promoter binding provides new insight into the physiological regulation of cell genes by EBNA-2.
Copyright © 2014, American Society for Microbiology. All Rights Reserved.
Figures

References
-
- White RE, Ramer PC, Naresh KN, Meixlsperger S, Pinaud L, Rooney C, Savoldo B, Coutinho R, Bodor C, Gribben J, Ibrahim HA, Bower M, Nourse JP, Gandhi MK, Middeldorp J, Cader FZ, Murray P, Munz C, Allday MJ. 2012. EBNA3B-deficient EBV promotes B cell lymphomagenesis in humanized mice and is found in human tumors. J. Clin. Invest. 122:1487–1502. 10.1172/JCI58092 - DOI - PMC - PubMed
-
- Kieff E, Rickinson A. 2007. Epstein-Barr virus, p 2603–2654 In Knipe DM, Howley PM, Griffin DE, Lamb RA, Martin MA, Roizman B, Straus SE. (ed), Fields virology, 5th ed. Lippincott Williams & Wilkins, Philadelphia, PA
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
