

Structural insight into BH3 domain binding of vaccinia virus antiapoptotic F1L
- PMID: 24850748
- PMCID: PMC4135939
- DOI: 10.1128/JVI.01092-14
Structural insight into BH3 domain binding of vaccinia virus antiapoptotic F1L
Abstract
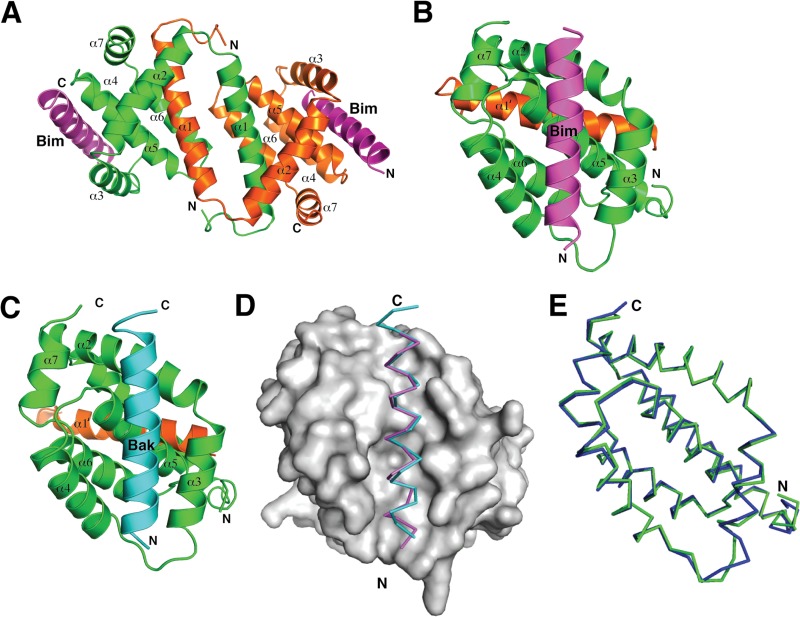
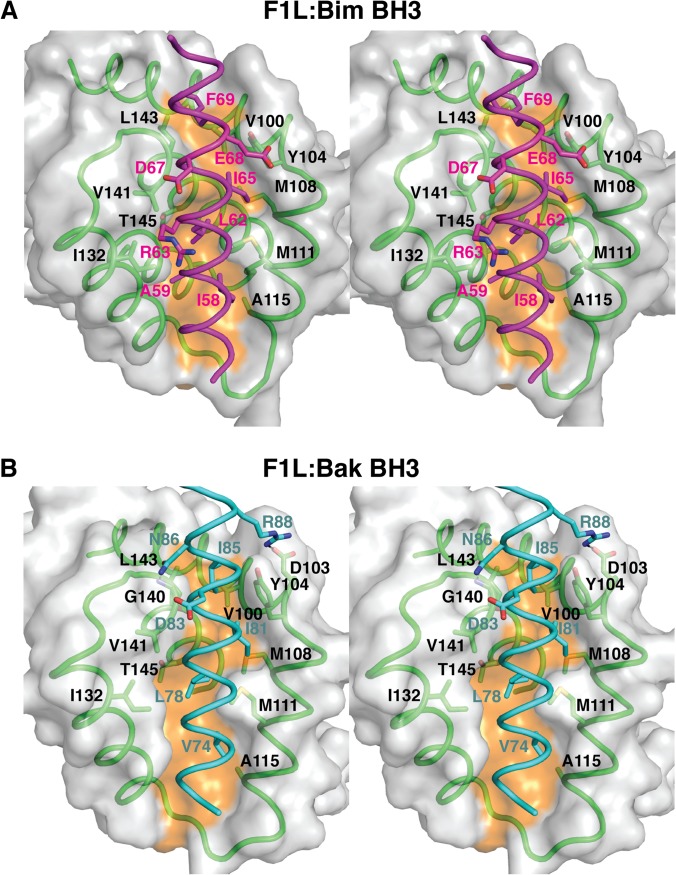
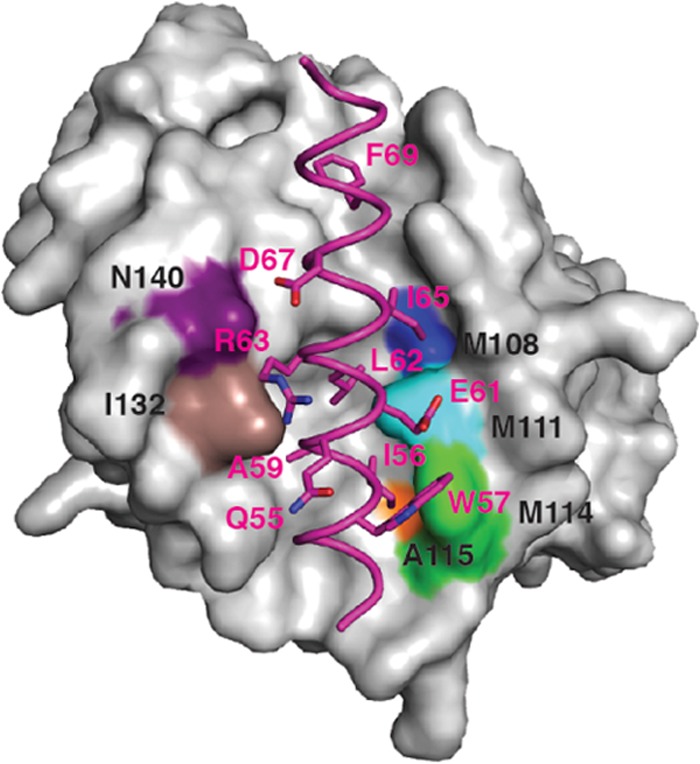
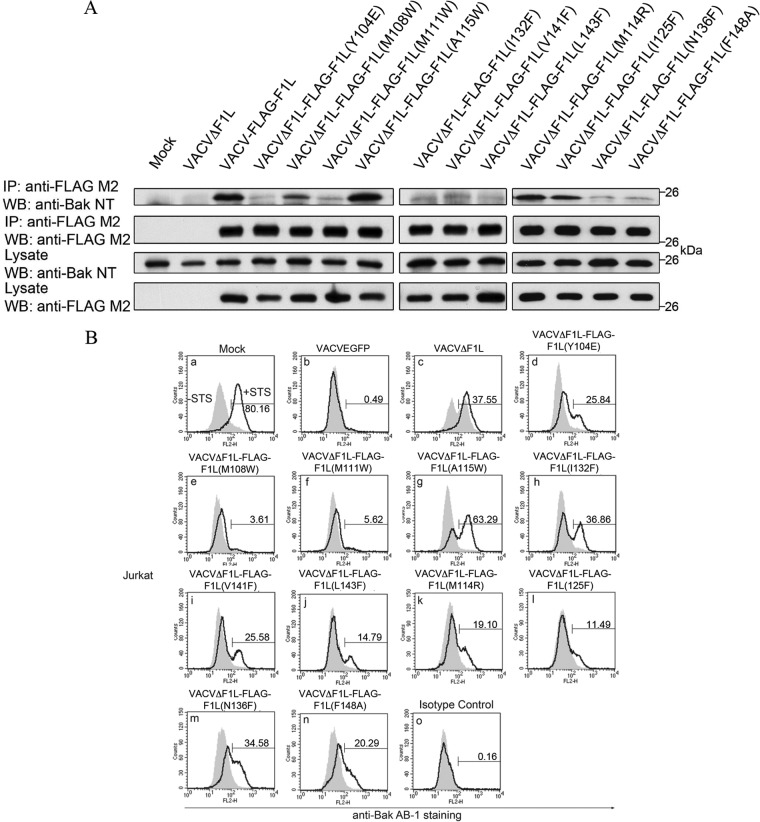
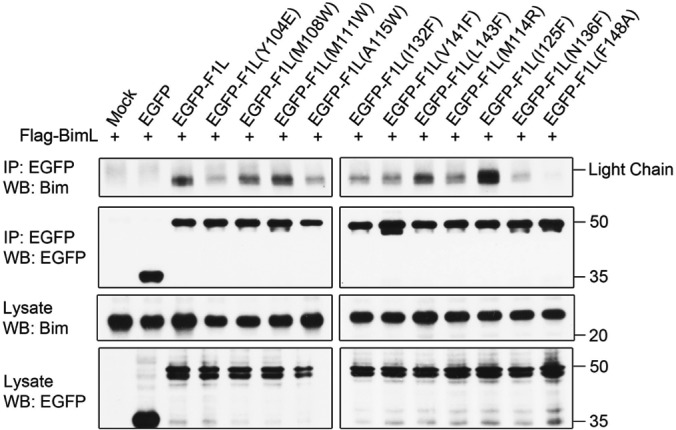
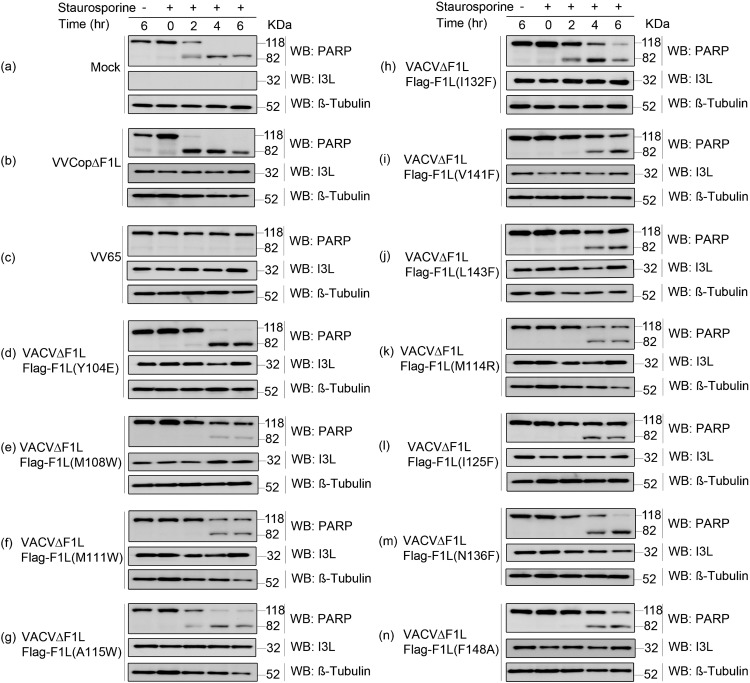
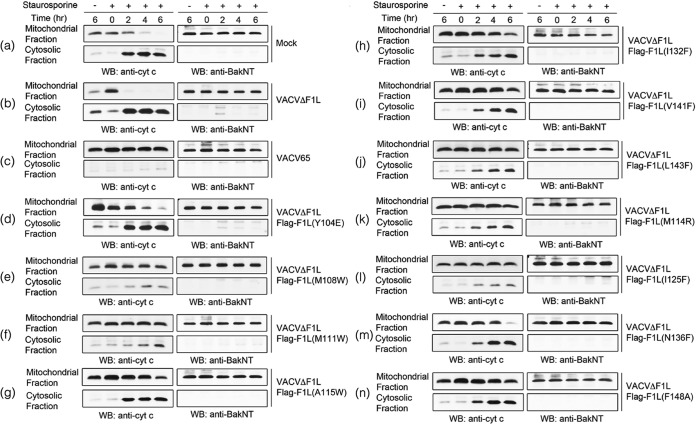
Apoptosis is a tightly regulated process that plays a crucial role in the removal of virus-infected cells, a process controlled by both pro- and antiapoptotic members of the Bcl-2 family. The proapoptotic proteins Bak and Bax are regulated by antiapoptotic Bcl-2 proteins and are also activated by a subset of proteins known as BH3-only proteins that perform dual functions by directly activating Bak and Bax or by sequestering and neutralizing antiapoptotic family members. Numerous viruses express proteins that prevent premature host cell apoptosis. Vaccinia virus encodes F1L, an antiapoptotic protein essential for survival of infected cells that bears no discernible sequence homology to mammalian cell death inhibitors. Despite the limited sequence similarities, F1L has been shown to adopt a novel dimeric Bcl-2-like fold that enables hetero-oligomeric binding to both Bak and the proapoptotic BH3-only protein Bim that ultimately prevents Bak and Bax homo-oligomerization. However, no structural data on the mode of engagement of F1L and its Bcl-2 counterparts are available. Here we solved the crystal structures of F1L in complex with two ligands, Bim and Bak. Our structures indicate that F1L can engage two BH3 ligands simultaneously via the canonical Bcl-2 ligand binding grooves. Furthermore, by structure-guided mutagenesis, we generated point mutations within the binding pocket of F1L in order to elucidate the residues responsible for both Bim and Bak binding and prevention of apoptosis. We propose that the sequestration of Bim by F1L is primarily responsible for preventing apoptosis during vaccinia virus infection.
Importance: Numerous viruses have adapted strategies to counteract apoptosis by encoding proteins responsible for sequestering proapoptotic components. Vaccinia virus, the prototypical member of the family Orthopoxviridae, encodes a protein known as F1L that functions to prevent apoptosis by interacting with Bak and the BH3-only protein Bim. Despite recent structural advances, little is known regarding the mechanics of binding between F1L and the proapoptotic Bcl-2 family members. Utilizing three-dimensional structures of F1L bound to host proapoptotic proteins, we generated variants of F1L that neutralize Bim and/or Bak. We demonstrate that during vaccinia virus infection, engagement of Bim and Bak by F1L is crucial for subversion of host cell apoptosis.
Copyright © 2014, American Society for Microbiology. All Rights Reserved.
Figures
References
Publication types
MeSH terms
Substances
Associated data
- Actions
- Actions
- Actions
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
