

Mobile elements drive recombination hotspots in the core genome of Staphylococcus aureus
- PMID: 24853639
- PMCID: PMC4036114
- DOI: 10.1038/ncomms4956
Mobile elements drive recombination hotspots in the core genome of Staphylococcus aureus
Abstract
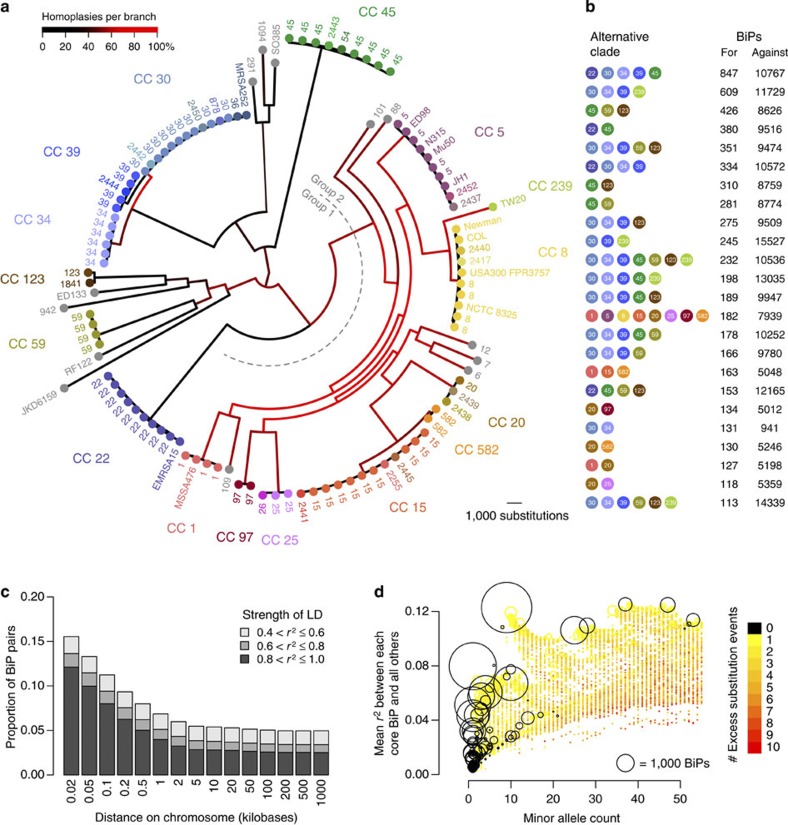
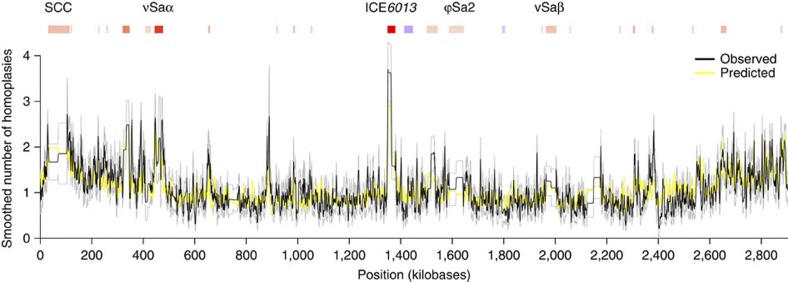
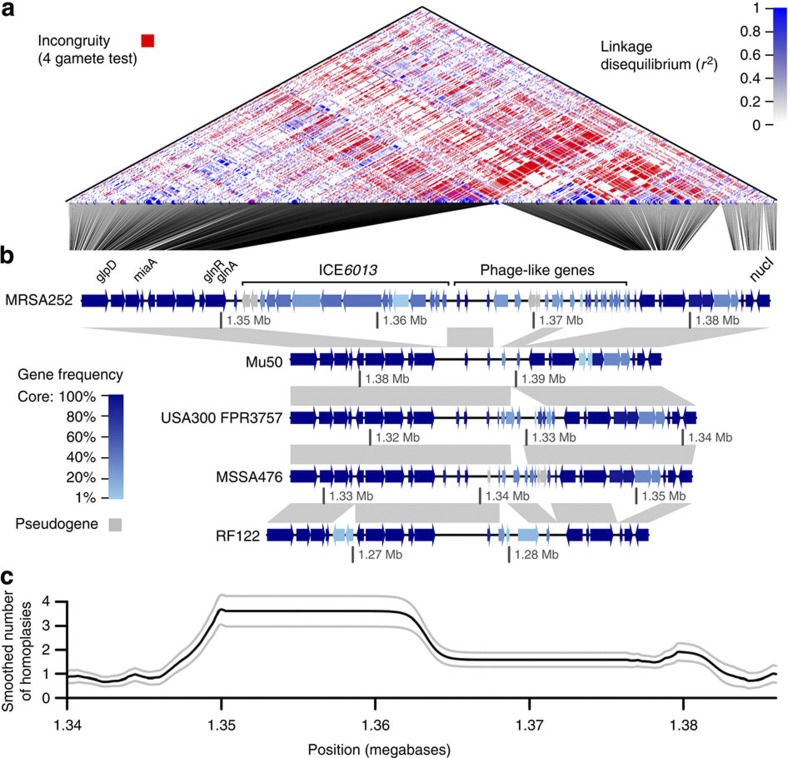
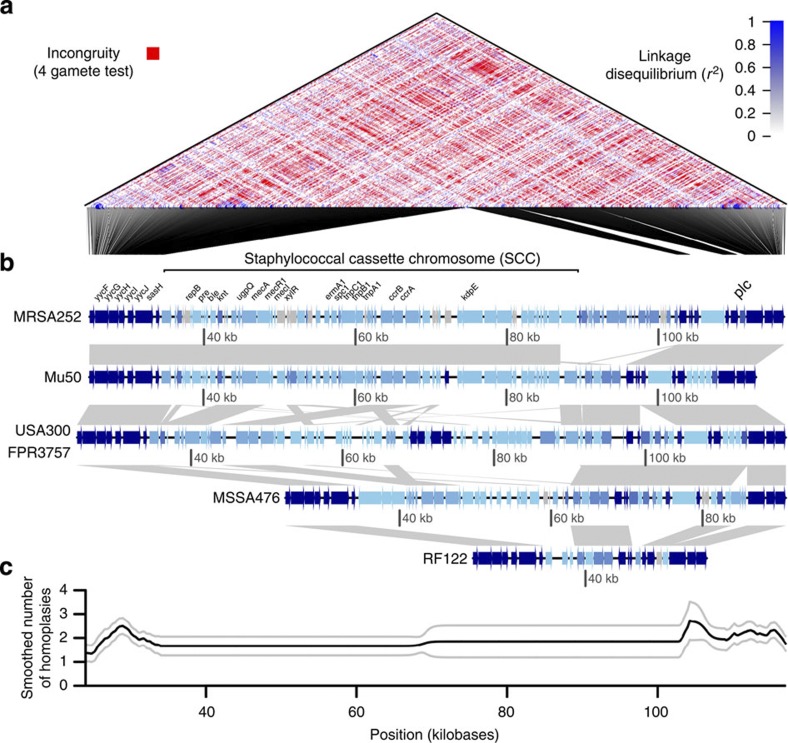
Horizontal gene transfer is an important driver of bacterial evolution, but genetic exchange in the core genome of clonal species, including the major pathogen Staphylococcus aureus, is incompletely understood. Here we reveal widespread homologous recombination in S. aureus at the species level, in contrast to its near-complete absence between closely related strains. We discover a patchwork of hotspots and coldspots at fine scales falling against a backdrop of broad-scale trends in rate variation. Over megabases, homoplasy rates fluctuate 1.9-fold, peaking towards the origin-of-replication. Over kilobases, we find core recombination hotspots of up to 2.5-fold enrichment situated near fault lines in the genome associated with mobile elements. The strongest hotspots include regions flanking conjugative transposon ICE6013, the staphylococcal cassette chromosome (SCC) and genomic island νSaα. Mobile element-driven core genome transfer represents an opportunity for adaptation and challenges our understanding of the recombination landscape in predominantly clonal pathogens, with important implications for genotype-phenotype mapping.
Figures
References
-
- Ochman H., Lawrence J. G. & Groisman E. A. Lateral gene transfer and the nature of bacterial innovation. Nature 405, 299–304 (2000). - PubMed
-
- Wright G. D. The antibiotic resistome: the nexus of chemical and genetic diversity. Nat. Rev. Microbiol. 5, 175–186 (2007). - PubMed
-
- MacLean R. C., Hall A. R., Perron G. G. & Buckling A. The population genetics of antibiotic resistance: integrating molecular mechanisms and treatment contexts. Nat. Rev. Genet. 11, 405–414 (2010). - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
