

Improving mutation screening in familial hematuric nephropathies through next generation sequencing
- PMID: 24854265
- PMCID: PMC4243343
- DOI: 10.1681/ASN.2013080912
Improving mutation screening in familial hematuric nephropathies through next generation sequencing
Abstract
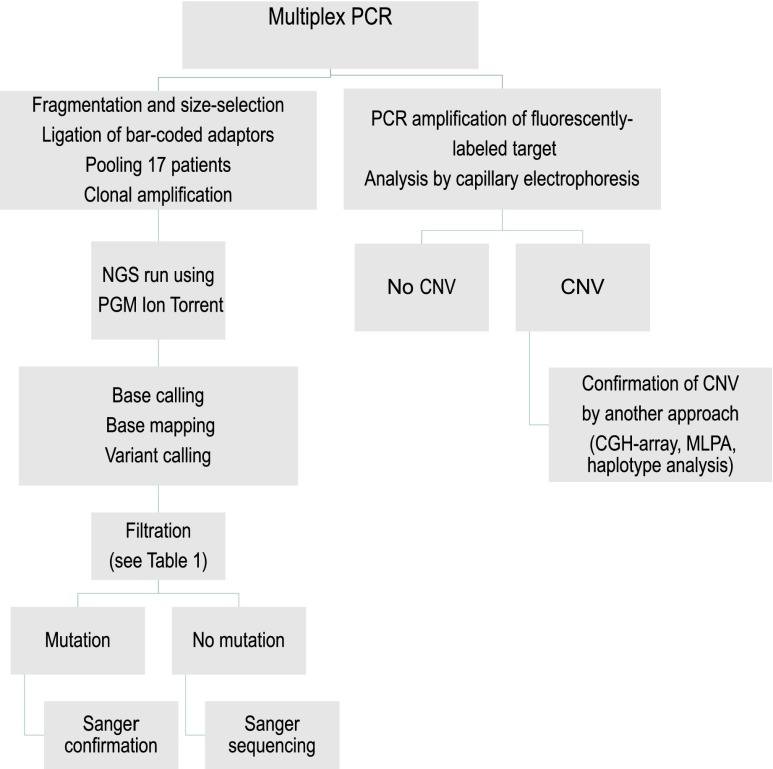
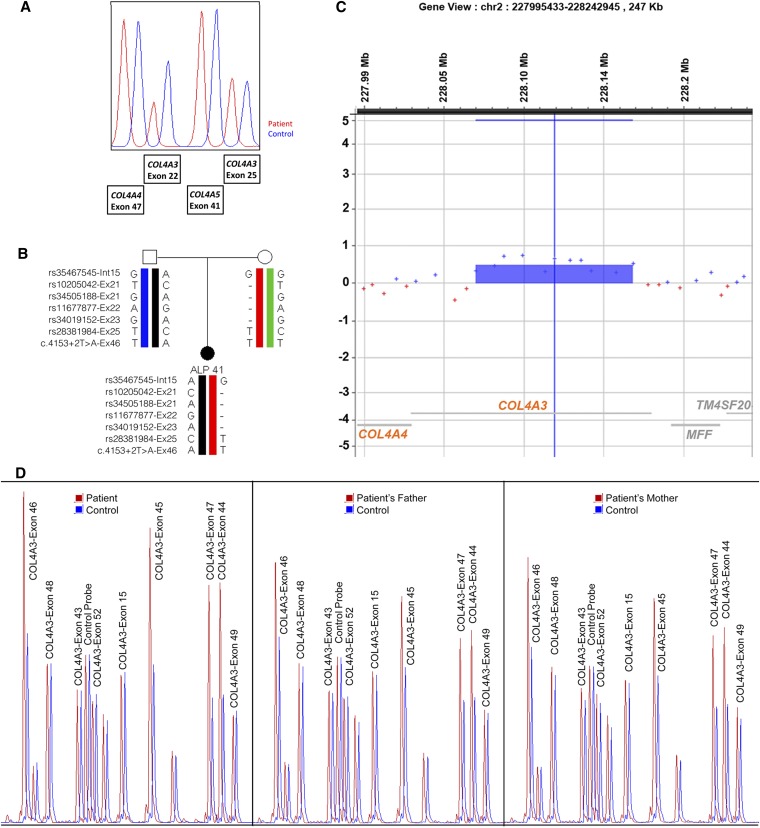
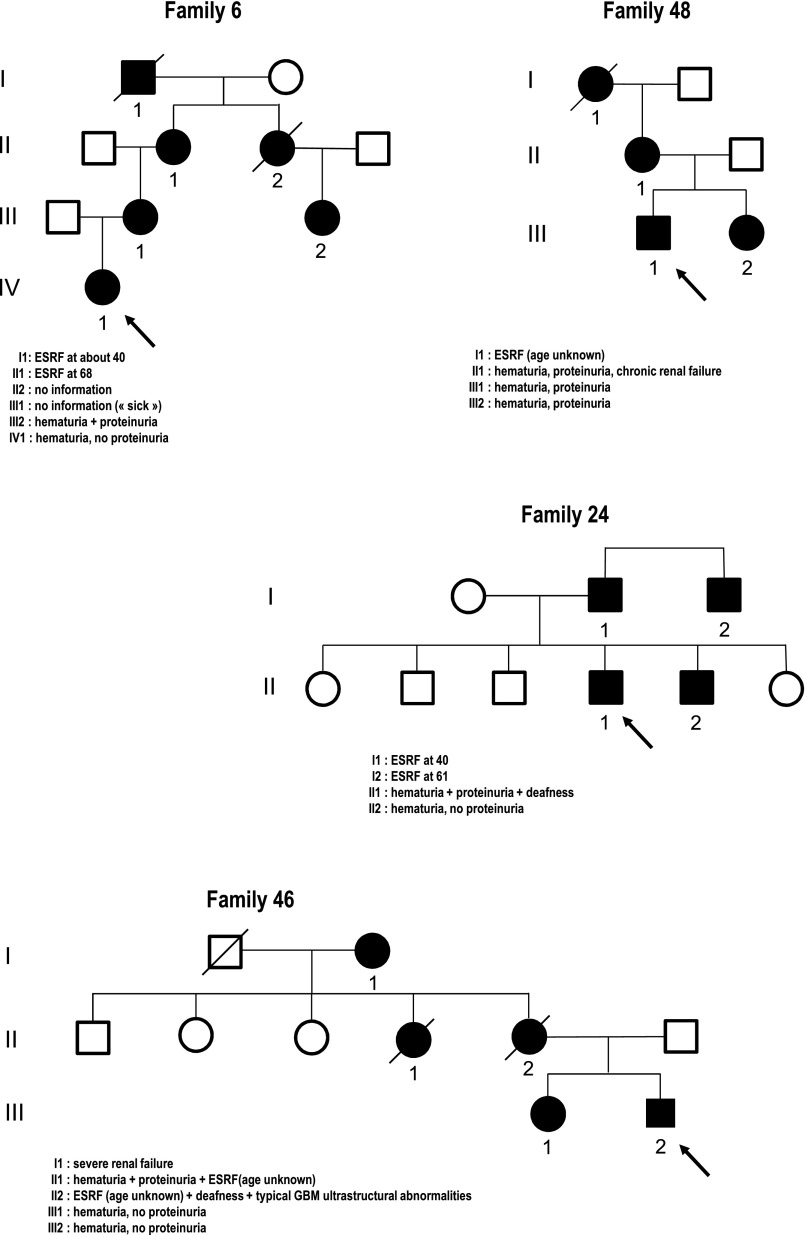
Alport syndrome is an inherited nephropathy associated with mutations in genes encoding type IV collagen chains present in the glomerular basement membrane. COL4A5 mutations are associated with the major X-linked form of the disease, and COL4A3 and COL4A4 mutations are associated with autosomal recessive and dominant forms (thought to be involved in 15% and 1%-5% of the families, respectively) and benign familial hematuria. Mutation screening of these three large genes is time-consuming and expensive. Here, we carried out a combination of multiplex PCR, amplicon quantification, and next generation sequencing (NGS) analysis of three genes in 101 unrelated patients. We identified 88 mutations and 6 variations of unknown significance on 116 alleles in 83 patients. Two additional indel mutations were found only by secondary Sanger sequencing, but they were easily identified retrospectively with the web-based sequence visualization tool Integrative Genomics Viewer. Altogether, 75 mutations were novel. Sequencing the three genes simultaneously was particularly advantageous as the mode of inheritance could not be determined with certainty in many instances. The proportion of mutations in COL4A3 and COL4A4 was notably high, and the autosomal dominant forms of Alport syndrome appear more frequently than reported previously. Finally, this approach allowed the identification of large COL4A3 and COL4A4 rearrangements not described previously. We conclude that NGS is efficient, reduces screening time and cost, and facilitates the provision of appropriate genetic counseling in Alport syndrome.
Keywords: Alport syndrome; genetic renal disease; molecular genetics.
Copyright © 2014 by the American Society of Nephrology.
Figures
References
-
- Kashtan CE, Michael AF: Alport syndrome. Kidney Int 50: 1445–1463, 1996 - PubMed
-
- Gretz N, Broyer M, Brunner FP, Brynger H, Donckerwolcke RA, Jacobs C, Kramer P, Selwood NH, Wing AJ: Alport’s syndrome as a cause of renal failure in Europe. Pediatr Nephrol 1: 411–415, 1987 - PubMed
-
- Atkin CL, Gregory MC, Border WA: Alport syndrome. In: Diseases of the Kidney, edited by Schrier RW, Gottschalk CW, Boston, Little and Brown, 1988, pp 617–641
-
- Levy M, Feingold J: Estimating prevalence in single-gene kidney diseases progressing to renal failure. Kidney Int 58: 925–943, 2000 - PubMed
Publication types
MeSH terms
Substances
Supplementary concepts
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
