

MicroRNA-24 antagonism prevents renal ischemia reperfusion injury
- PMID: 24854275
- PMCID: PMC4243358
- DOI: 10.1681/ASN.2013121329
MicroRNA-24 antagonism prevents renal ischemia reperfusion injury
Abstract
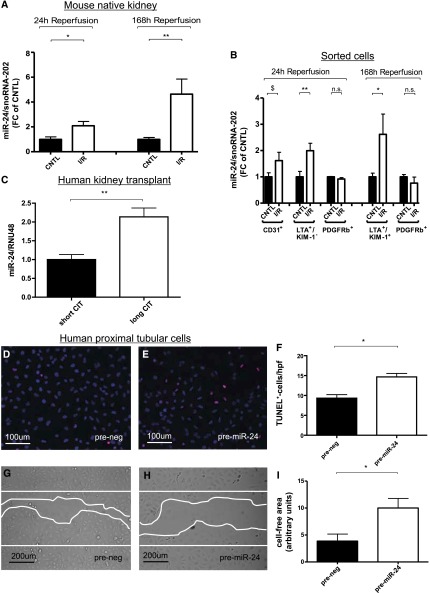
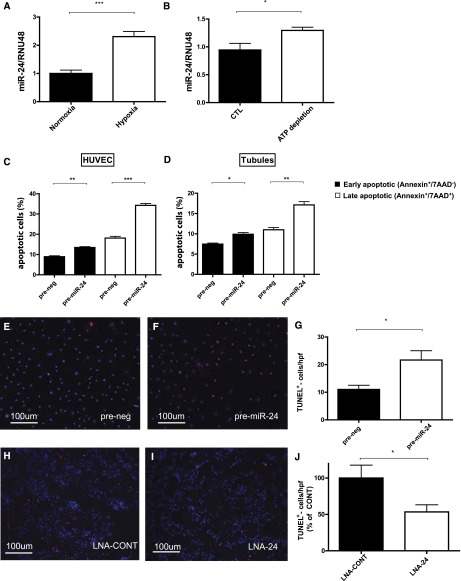
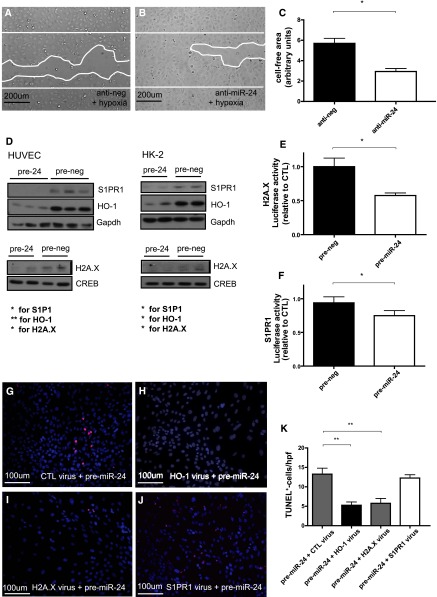
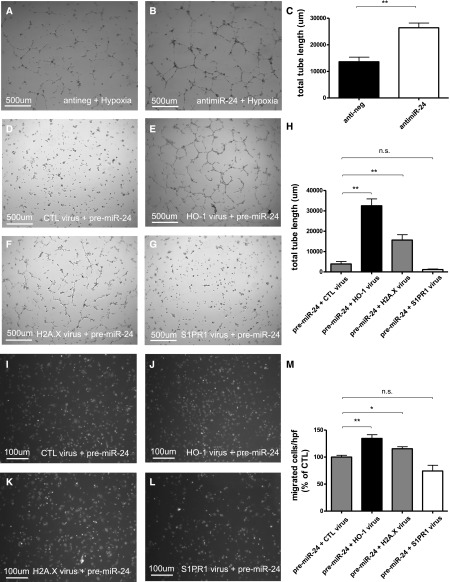
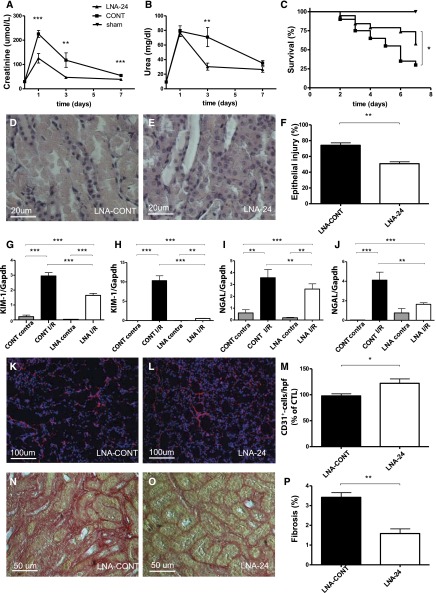
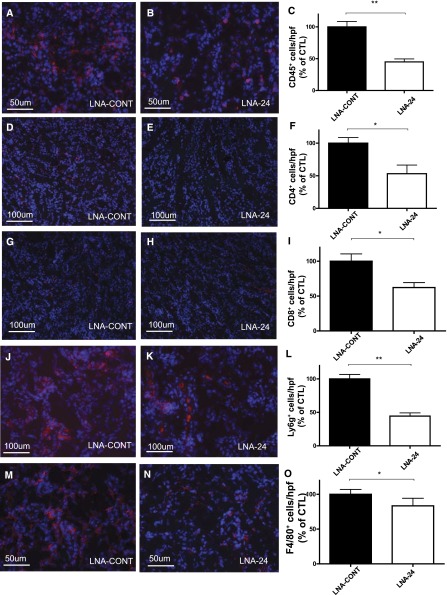
Ischemia-reperfusion (I/R) injury of the kidney is a major cause of AKI. MicroRNAs (miRs) are powerful regulators of various diseases. We investigated the role of apoptosis-associated miR-24 in renal I/R injury. miR-24 was upregulated in the kidney after I/R injury of mice and in patients after kidney transplantation. Cell-sorting experiments revealed a specific miR-24 enrichment in renal endothelial and tubular epithelial cells after I/R induction. In vitro, anoxia/hypoxia induced an enrichment of miR-24 in endothelial and tubular epithelial cells. Transient overexpression of miR-24 alone induced apoptosis and altered functional parameters in these cells, whereas silencing of miR-24 ameliorated apoptotic responses and rescued functional parameters in hypoxic conditions. miR-24 effects were mediated through regulation of H2A histone family, member X, and heme oxygenase 1, which were experimentally validated as direct miR-24 targets through luciferase reporter assays. In vitro, adenoviral overexpression of miR-24 targets lacking miR-24 binding sites along with miR-24 precursors rescued various functional parameters in endothelial and tubular epithelial cells. In vivo, silencing of miR-24 in mice before I/R injury resulted in a significant improvement in survival and kidney function, a reduction of apoptosis, improved histologic tubular epithelial injury, and less infiltration of inflammatory cells. miR-24 also regulated heme oxygenase 1 and H2A histone family, member X, in vivo. Overall, these results indicate miR-24 promotes renal ischemic injury by stimulating apoptosis in endothelial and tubular epithelial cell. Therefore, miR-24 inhibition may be a promising future therapeutic option in the treatment of patients with ischemic AKI.
Keywords: acute renal failure; apoptosis; endothelium; heme oxygenase; ischemia-reperfusion; proximal tubule.
Copyright © 2014 by the American Society of Nephrology.
Figures

References
-
- Kelly KJ: Acute renal failure: Much more than a kidney disease. Semin Nephrol 26: 105–113, 2006 - PubMed
-
- Bon D, Chatauret N, Giraud S, Thuillier R, Favreau F, Hauet T: New strategies to optimize kidney recovery and preservation in transplantation. Nat Rev Nephrol 8: 339–347, 2012 - PubMed
-
- Weight SC, Bell PR, Nicholson ML: Renal ischaemia—reperfusion injury. Br J Surg 83: 162–170, 1996 - PubMed
-
- Lorenzen JM, Haller H, Thum T: MicroRNAs as mediators and therapeutic targets in chronic kidney disease. Nat Rev Nephrol 7: 286–294, 2011 - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
