

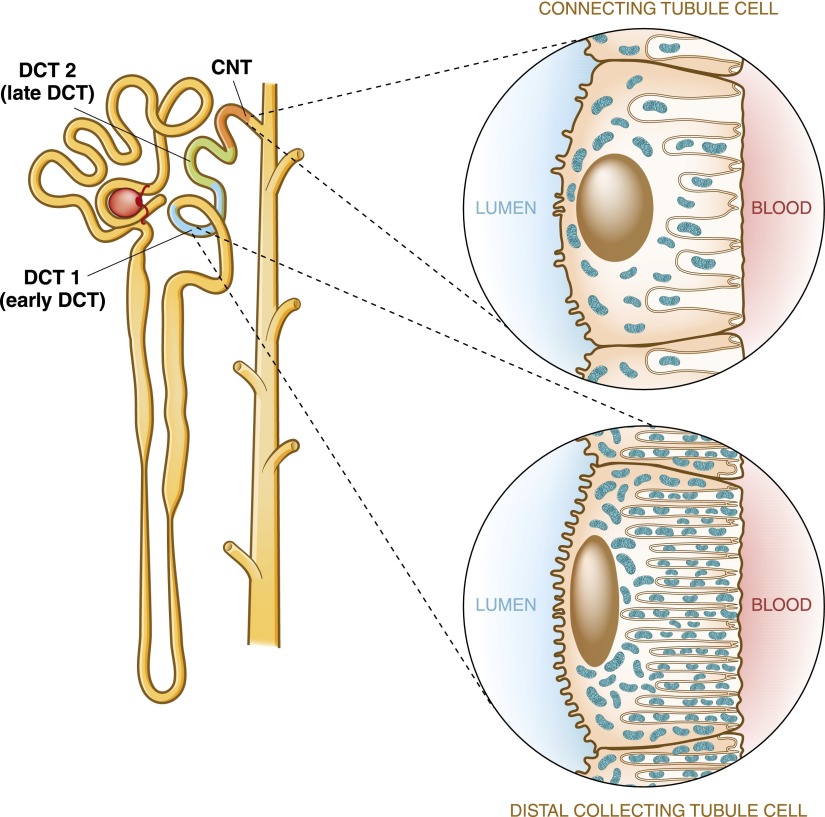
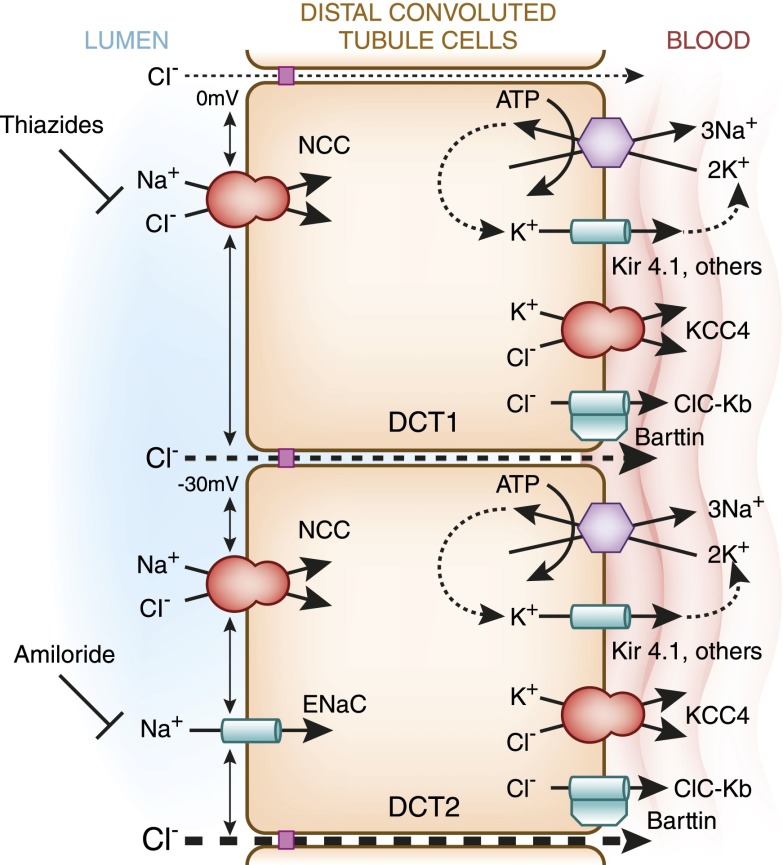
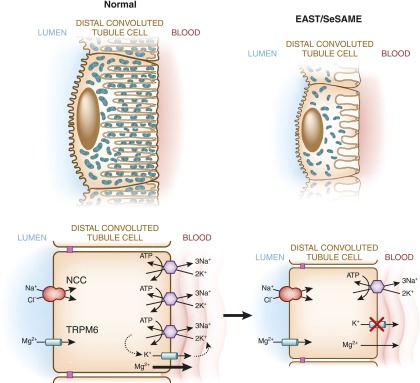
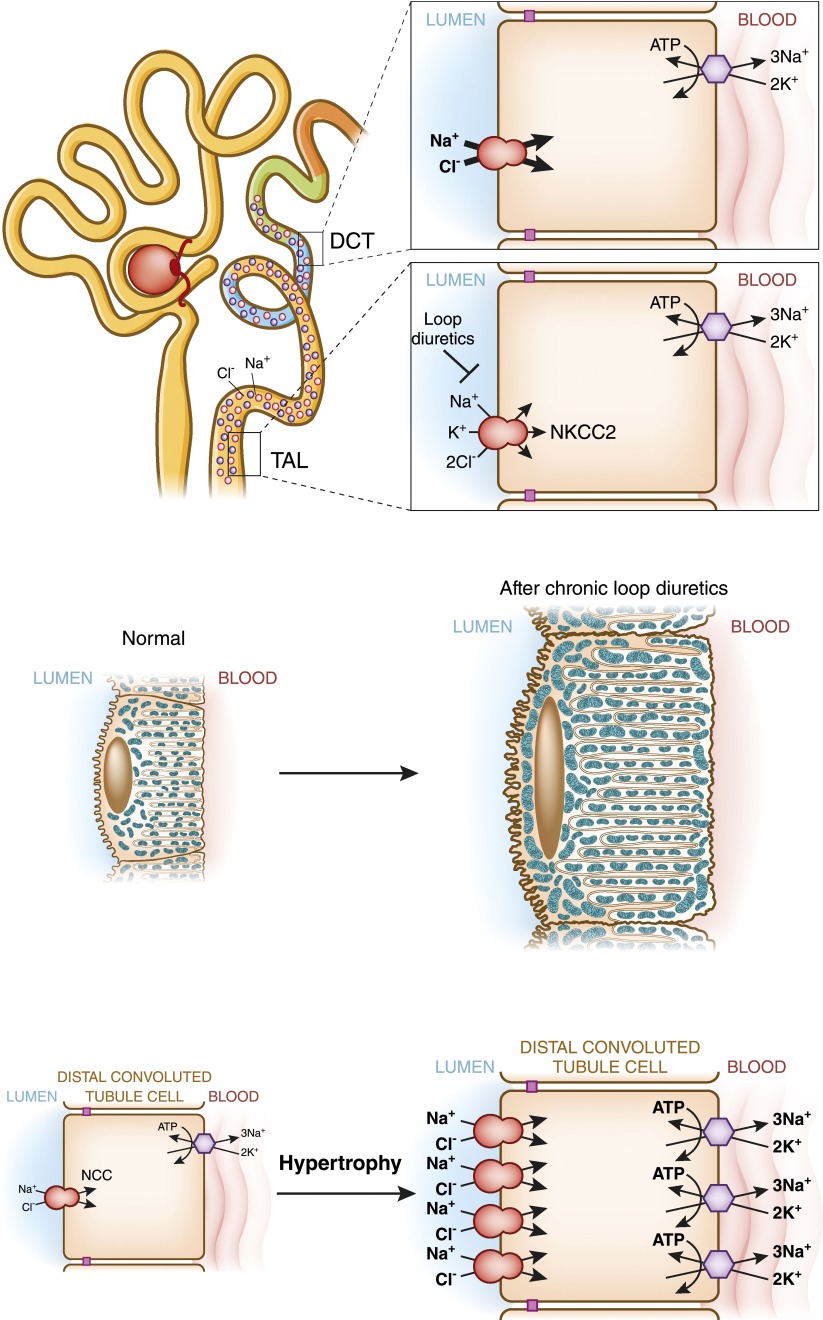
Distal convoluted tubule
- PMID: 24855283
- PMCID: PMC4255408
- DOI: 10.2215/CJN.05920613
Distal convoluted tubule
Abstract
The distal convoluted tubule is the nephron segment that lies immediately downstream of the macula densa. Although short in length, the distal convoluted tubule plays a critical role in sodium, potassium, and divalent cation homeostasis. Recent genetic and physiologic studies have greatly expanded our understanding of how the distal convoluted tubule regulates these processes at the molecular level. This article provides an update on the distal convoluted tubule, highlighting concepts and pathophysiology relevant to clinical practice.
Keywords: Na transport; distal tubule; mineral metabolism; potassium channels; renal physiology.
Copyright © 2014 by the American Society of Nephrology.
Figures
References
-
- Reilly RF, Ellison DH: Mammalian distal tubule: Physiology, pathophysiology, and molecular anatomy. Physiol Rev 80: 277–313, 2000 - PubMed
-
- Arriza JL, Weinberger C, Cerelli G, Glaser TM, Handelin BL, Housman DE, Evans RM: Cloning of human mineralocorticoid receptor complementary DNA: Structural and functional kinship with the glucocorticoid receptor. Science 237: 268–275, 1987 - PubMed
-
- Bostanjoglo M, Reeves WB, Reilly RF, Velázquez H, Robertson N, Litwack G, Morsing P, Dørup J, Bachmann S, Ellison DH: 11Beta-hydroxysteroid dehydrogenase, mineralocorticoid receptor, and thiazide-sensitive Na-Cl cotransporter expression by distal tubules. J Am Soc Nephrol 9: 1347–1358, 1998 - PubMed
-
- Meneton P, Loffing J, Warnock DG: Sodium and potassium handling by the aldosterone-sensitive distal nephron: The pivotal role of the distal and connecting tubule. Am J Physiol Renal Physiol 287: F593–F601, 2004 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
