

A roadmap for functional structural variants in the soybean genome
- PMID: 24855315
- PMCID: PMC4455779
- DOI: 10.1534/g3.114.011551
A roadmap for functional structural variants in the soybean genome
Abstract
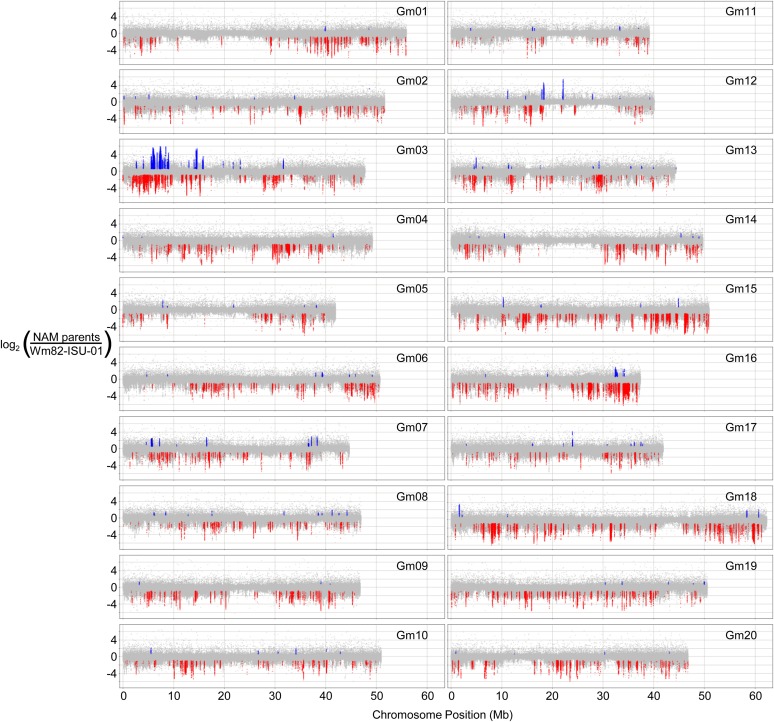
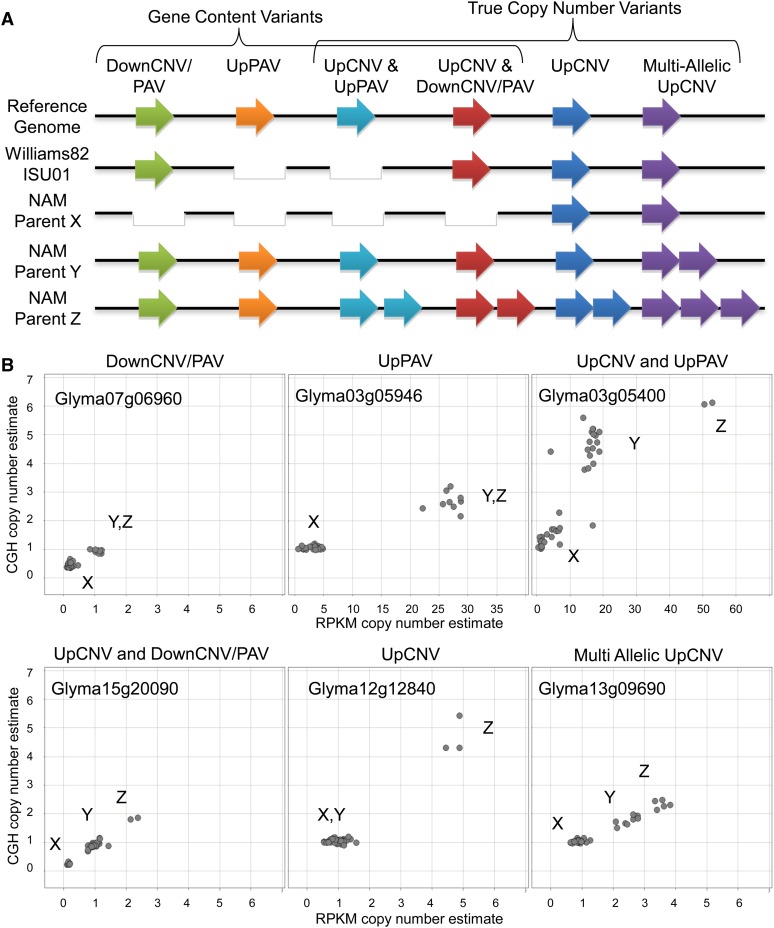
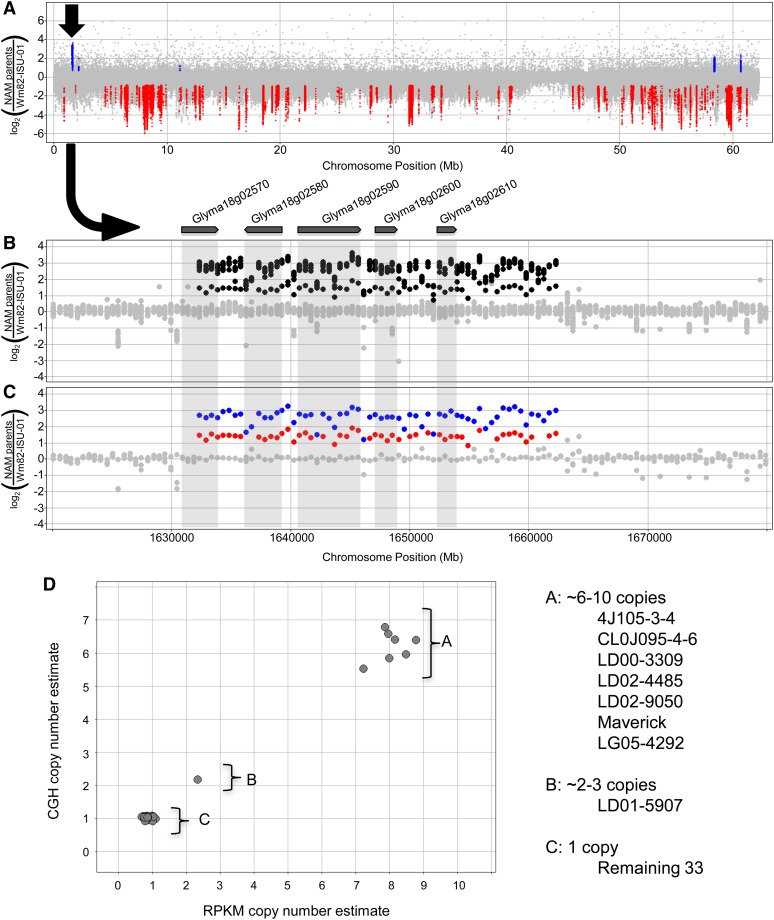
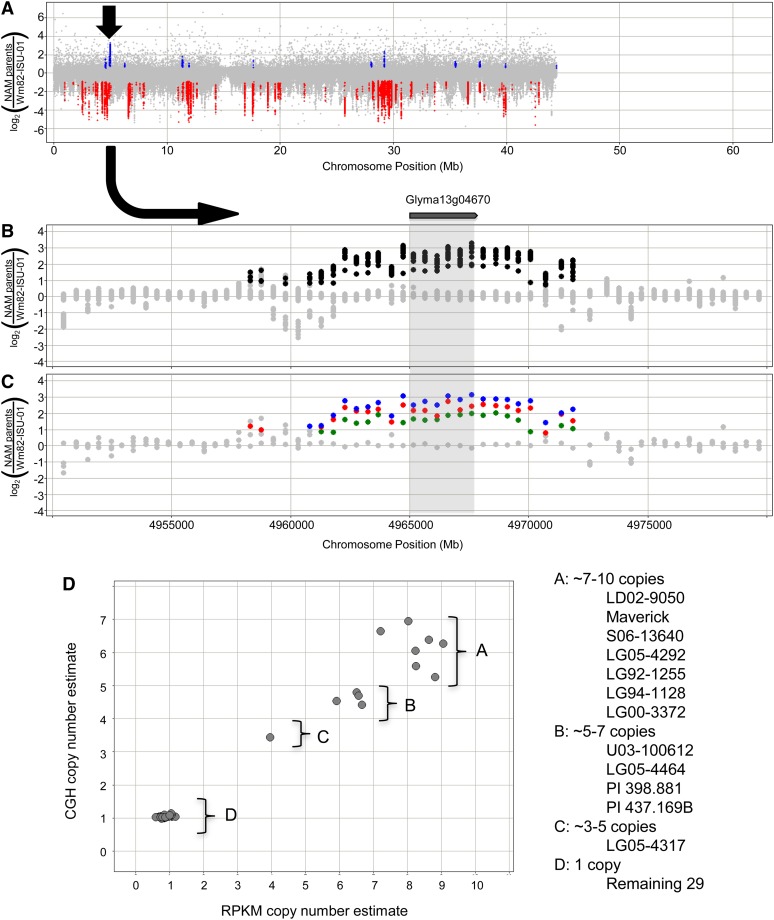
Gene structural variation (SV) has recently emerged as a key genetic mechanism underlying several important phenotypic traits in crop species. We screened a panel of 41 soybean (Glycine max) accessions serving as parents in a soybean nested association mapping population for deletions and duplications in more than 53,000 gene models. Array hybridization and whole genome resequencing methods were used as complementary technologies to identify SV in 1528 genes, or approximately 2.8%, of the soybean gene models. Although SV occurs throughout the genome, SV enrichment was noted in families of biotic defense response genes. Among accessions, SV was nearly eightfold less frequent for gene models that have retained paralogs since the last whole genome duplication event, compared with genes that have not retained paralogs. Increases in gene copy number, similar to that described at the Rhg1 resistance locus, account for approximately one-fourth of the genic SV events. This assessment of soybean SV occurrence presents a target list of genes potentially responsible for rapidly evolving and/or adaptive traits.
Keywords: CNV; Glycine max; nested association mapping; soybean; structural variation.
Copyright © 2014 Anderson et al.
Figures
References
-
- Bernard R., Cremeens C., 1988. Registration of ‘Williams 82’soybean. Crop Sci. 28: 1027–1028.
-
- Cao J., Schneeberger K., Ossowski S., Günther T., Bender S., et al. , 2011. Whole-genome sequencing of multiple Arabidopsis thaliana populations. Nat. Genet. 43: 956–963. - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
- SRA/SRP042221
- SRA/SRP042236
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases