

Rare inherited kidney diseases: challenges, opportunities, and perspectives
- PMID: 24856029
- PMCID: PMC4135047
- DOI: 10.1016/S0140-6736(14)60659-0
Rare inherited kidney diseases: challenges, opportunities, and perspectives
Abstract
At least 10% of adults and nearly all children who receive renal-replacement therapy have an inherited kidney disease. These patients rarely die when their disease progresses and can remain alive for many years because of advances in organ-replacement therapy. However, these disorders substantially decrease their quality of life and have a large effect on health-care systems. Since the kidneys regulate essential homoeostatic processes, inherited kidney disorders have multisystem complications, which add to the usual challenges for rare disorders. In this review, we discuss the nature of rare inherited kidney diseases, the challenges they pose, and opportunities from technological advances, which are well suited to target the kidney. Mechanistic insights from rare disorders are relevant for common disorders such as hypertension, kidney stones, cardiovascular disease, and progression of chronic kidney disease.
Copyright © 2014 Elsevier Ltd. All rights reserved.
Figures
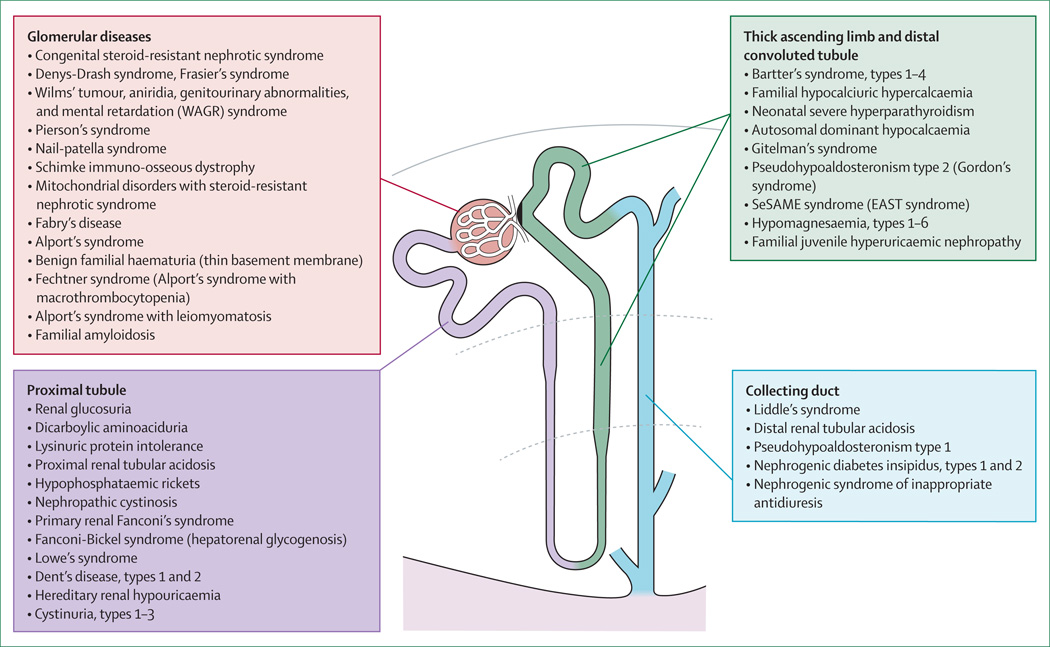
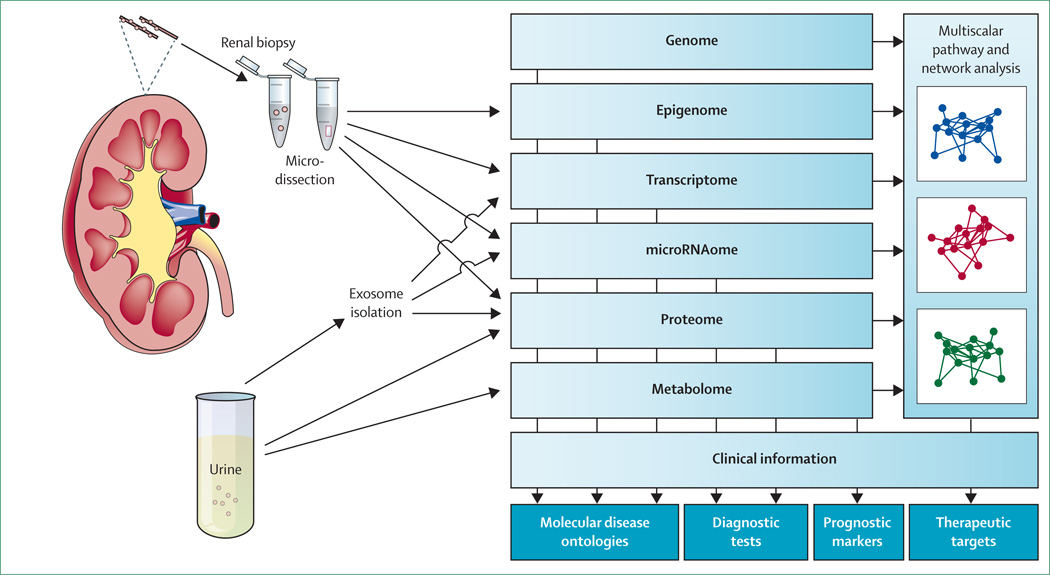
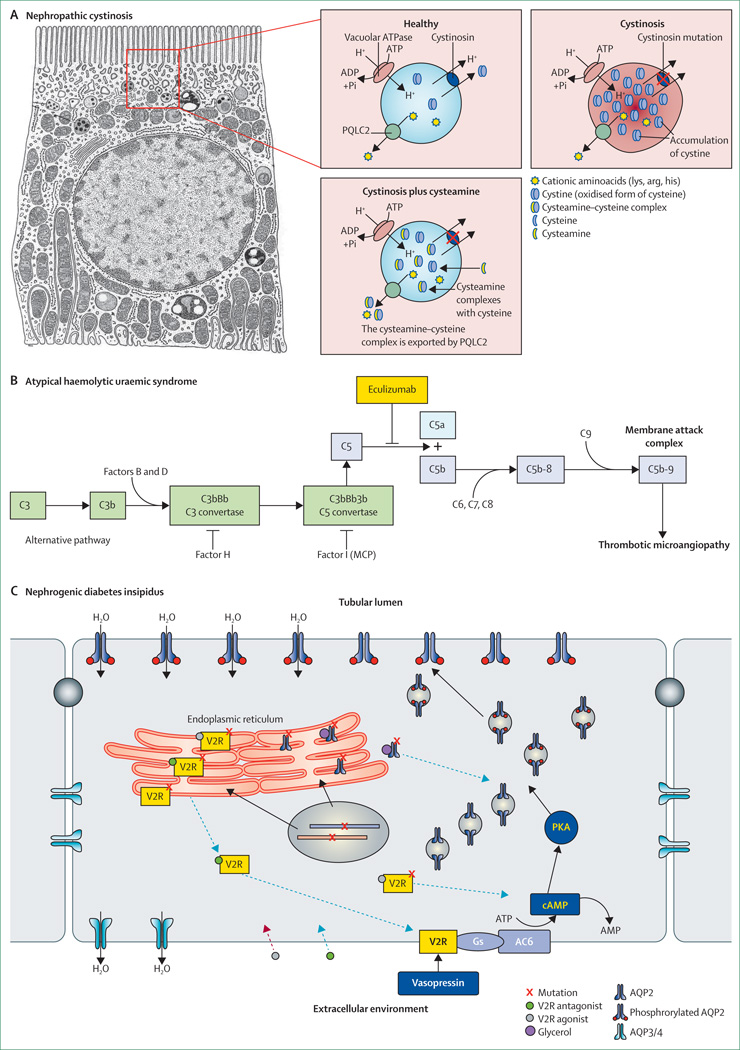
References
-
- Schieppati A, Henter JI, Daina E, Aperia A. Why rare diseases are an important medical and social issue. Lancet. 2008;371:2039–2041. - PubMed
-
- Hayashi S, Umeda T. 35 years of Japanese policy on rare diseases. Lancet. 2008;372:889–890. - PubMed
-
- Liu BC, He L, He G, He Y. A cross-national comparative study of orphan drug policies in the United States, the European Union, and Japan: towards a made-in-China orphan drug policy. J Public Health Policy. 2010;31:407–420. - PubMed
-
- Soliman NA. Orphan Kidney Diseases. Nephron Clin Pract. 2012;120:c194–c199. - PubMed
-
- [accessed March 31, 2014];Orphanet Report Series: Prevalence of rare diseases. 2013 Nov; http://www.orpha.net/orphacom/cahiers/docs/GB/Prevalence_of_rare_disease....
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
