

Pulmonary hypertension in wild type mice and animals with genetic deficit in KCa2.3 and KCa3.1 channels
- PMID: 24858807
- PMCID: PMC4032241
- DOI: 10.1371/journal.pone.0097687
Pulmonary hypertension in wild type mice and animals with genetic deficit in KCa2.3 and KCa3.1 channels
Abstract
Objective: In vascular biology, endothelial KCa2.3 and KCa3.1 channels contribute to arterial blood pressure regulation by producing membrane hyperpolarization and smooth muscle relaxation. The role of KCa2.3 and KCa3.1 channels in the pulmonary circulation is not fully established. Using mice with genetically encoded deficit of KCa2.3 and KCa3.1 channels, this study investigated the effect of loss of the channels in hypoxia-induced pulmonary hypertension.
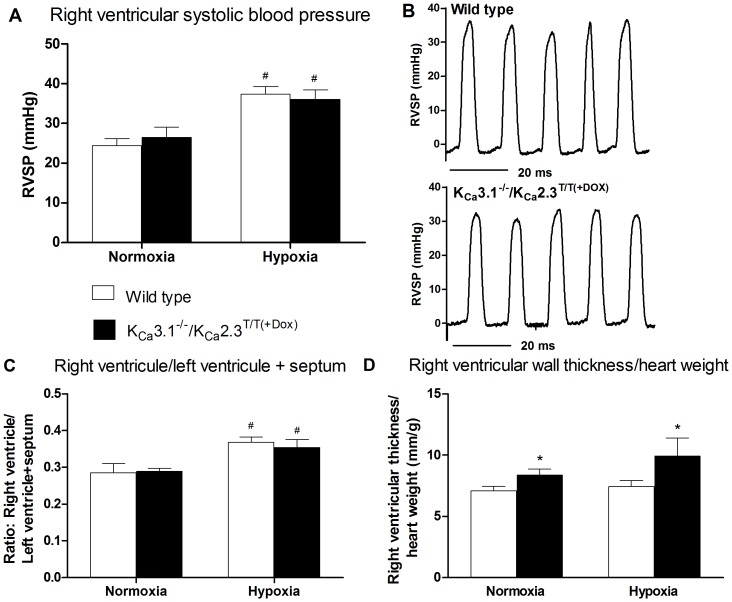
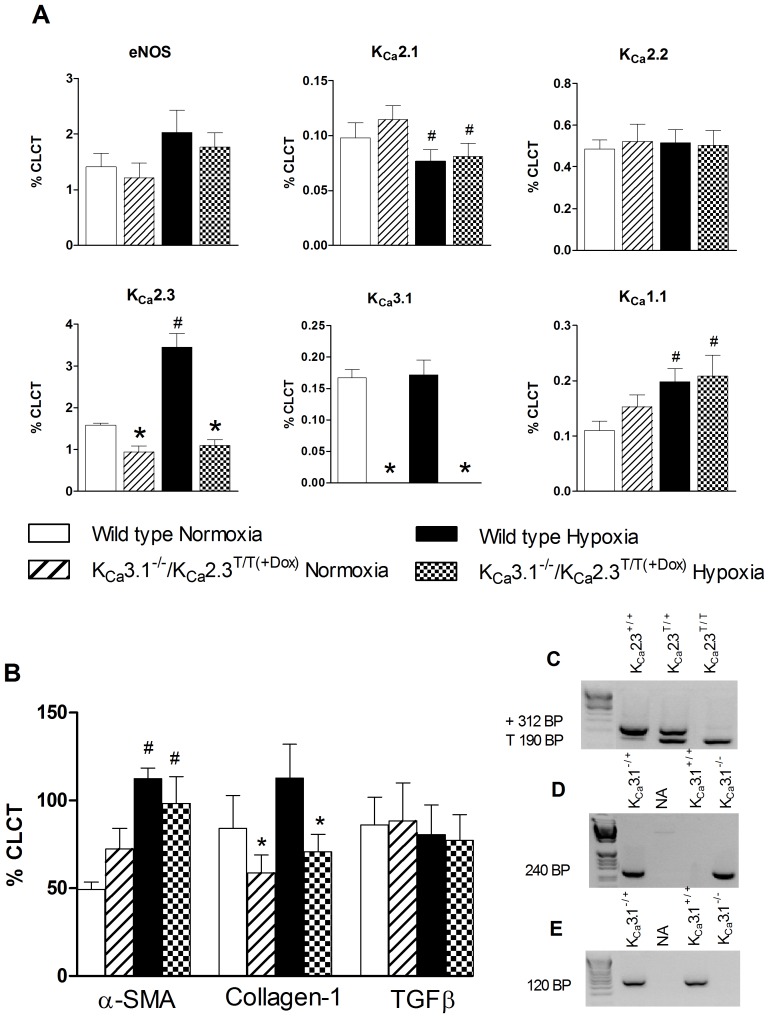
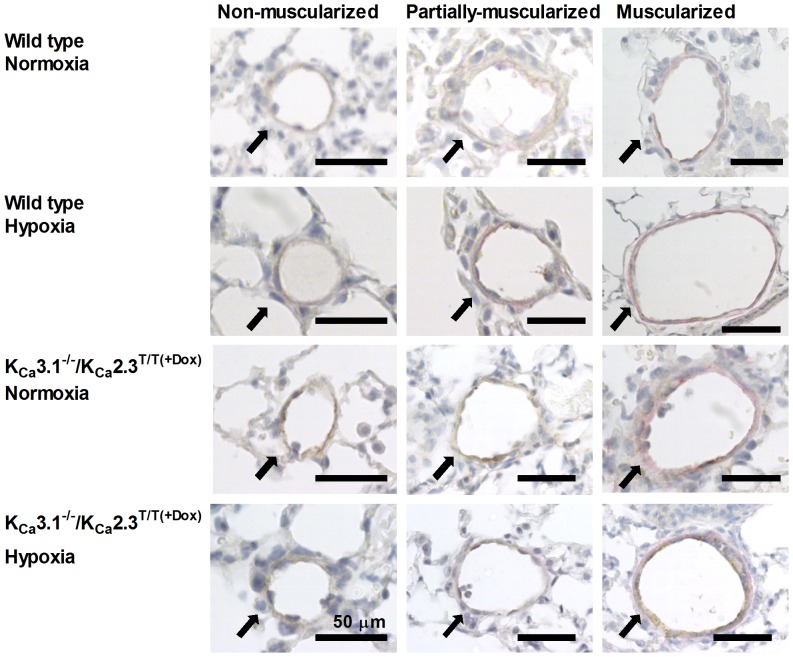
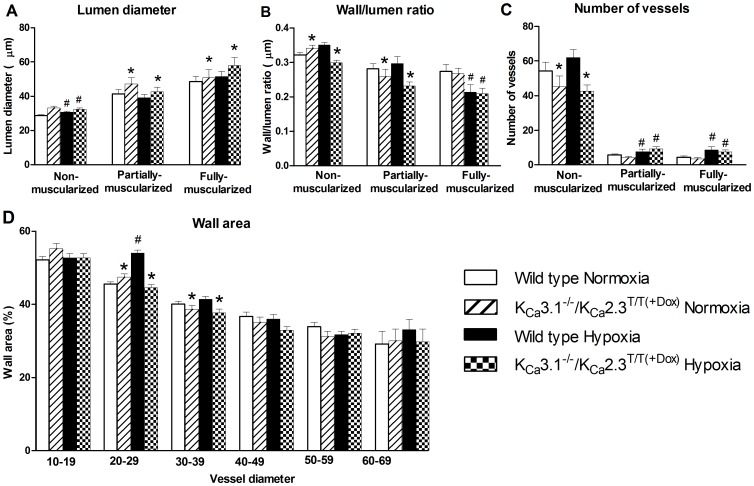
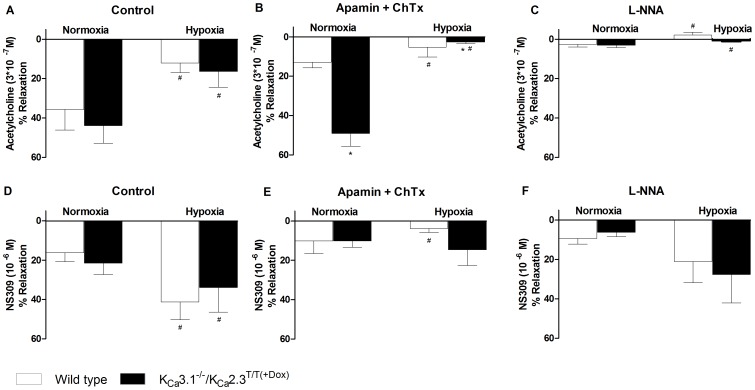
Approach and result: Male wild type and KCa3.1-/-/KCa2.3T/T(+DOX) mice were exposed to chronic hypoxia for four weeks to induce pulmonary hypertension. The degree of pulmonary hypertension was evaluated by right ventricular pressure and assessment of right ventricular hypertrophy. Segments of pulmonary arteries were mounted in a wire myograph for functional studies and morphometric studies were performed on lung sections. Chronic hypoxia induced pulmonary hypertension, right ventricular hypertrophy, increased lung weight, and increased hematocrit levels in either genotype. The KCa3.1-/-/KCa2.3T/T(+DOX) mice developed structural alterations in the heart with increased right ventricular wall thickness as well as in pulmonary vessels with increased lumen size in partially- and fully-muscularized vessels and decreased wall area, not seen in wild type mice. Exposure to chronic hypoxia up-regulated the gene expression of the KCa2.3 channel by twofold in wild type mice and increased by 2.5-fold the relaxation evoked by the KCa2.3 and KCa3.1 channel activator NS309, whereas the acetylcholine-induced relaxation - sensitive to the combination of KCa2.3 and KCa3.1 channel blockers, apamin and charybdotoxin - was reduced by 2.5-fold in chronic hypoxic mice of either genotype.
Conclusion: Despite the deficits of the KCa2.3 and KCa3.1 channels failed to change hypoxia-induced pulmonary hypertension, the up-regulation of KCa2.3-gene expression and increased NS309-induced relaxation in wild-type mice point to a novel mechanism to counteract pulmonary hypertension and to a potential therapeutic utility of KCa2.3/KCa3.1 activators for the treatment of pulmonary hypertension.
Conflict of interest statement
Figures
Similar articles
-
The influence of DOCA-salt hypertension and chronic administration of the FAAH inhibitor URB597 on KCa2.3/KCa3.1-EDH-type relaxation in rat small mesenteric arteries.Vascul Pharmacol. 2017 Dec;99:65-73. doi: 10.1016/j.vph.2017.10.001. Epub 2017 Oct 14. Vascul Pharmacol. 2017. PMID: 29038048
-
Endothelial KCa3.1 and KCa2.3 Mediate S1P (Sphingosine-1-Phosphate)-Dependent Vasodilation and Blood Pressure Homeostasis.Arterioscler Thromb Vasc Biol. 2023 May;43(5):726-738. doi: 10.1161/ATVBAHA.122.318820. Epub 2023 Mar 23. Arterioscler Thromb Vasc Biol. 2023. PMID: 36951065
-
Upregulating vascular endothelial KCa2.3 channels alleviates pulmonary hypertension in mice.Mol Pharmacol. 2025 Jul;107(7):100048. doi: 10.1016/j.molpha.2025.100048. Epub 2025 May 20. Mol Pharmacol. 2025. PMID: 40532274
-
Channelopathy of small- and intermediate-conductance Ca2+-activated K+ channels.Acta Pharmacol Sin. 2023 Feb;44(2):259-267. doi: 10.1038/s41401-022-00935-1. Epub 2022 Jun 17. Acta Pharmacol Sin. 2023. PMID: 35715699 Free PMC article. Review.
-
Pharmacology of Small- and Intermediate-Conductance Calcium-Activated Potassium Channels.Annu Rev Pharmacol Toxicol. 2020 Jan 6;60:219-240. doi: 10.1146/annurev-pharmtox-010919-023420. Epub 2019 Jul 23. Annu Rev Pharmacol Toxicol. 2020. PMID: 31337271 Free PMC article. Review.
Cited by
-
Docosahexaenoic acid causes rapid pulmonary arterial relaxation via KCa channel-mediated hyperpolarisation in pulmonary hypertension.Eur Respir J. 2016 Oct;48(4):1127-1136. doi: 10.1183/13993003.01814-2015. Epub 2016 Aug 18. Eur Respir J. 2016. PMID: 27540020 Free PMC article.
-
Down-regulation of KCa2.3 channels causes erectile dysfunction in mice.Sci Rep. 2017 Jun 19;7(1):3839. doi: 10.1038/s41598-017-04188-5. Sci Rep. 2017. PMID: 28630432 Free PMC article.
-
Genetic deficit of KCa 3.1 channels protects against pulmonary circulatory collapse induced by TRPV4 channel activation.Br J Pharmacol. 2015 Sep;172(18):4493-4505. doi: 10.1111/bph.13234. Epub 2015 Jul 24. Br J Pharmacol. 2015. PMID: 26102209 Free PMC article.
-
Effect of Acetazolamide and Zoledronate on Simulated High Altitude-Induced Bone Loss.Front Endocrinol (Lausanne). 2022 Feb 9;13:831369. doi: 10.3389/fendo.2022.831369. eCollection 2022. Front Endocrinol (Lausanne). 2022. PMID: 35222286 Free PMC article.
-
Chronic Hypoxia Decreases Endothelial Connexin 40, Attenuates Endothelium-Dependent Hyperpolarization-Mediated Relaxation in Small Distal Pulmonary Arteries, and Leads to Pulmonary Hypertension.J Am Heart Assoc. 2020 Dec 15;9(24):e018327. doi: 10.1161/JAHA.120.018327. Epub 2020 Dec 12. J Am Heart Assoc. 2020. PMID: 33307937 Free PMC article.
References
-
- Benza RL, Miller DP, Barst RJ, Badesch DB, Frost AE, et al. (2012) An evaluation of long-term survival from time of diagnosis in pulmonary arterial hypertension from the REVEAL Registry. Chest 142: 448–456. - PubMed
-
- Ignarro LJ, Burke TM, Wood KS, Wolin MS, Kadowitz PJ (1984) Association between cyclic GMP accumulation and acetylcholine-elicited relaxation of bovine intrapulmonary artery. J Pharmacol Exp Ther 228: 682–690. - PubMed
-
- Palmer RM, Ferrige AG, Moncada S (1987) Nitric oxide release accounts for the biological activity of endothelium-derived relaxing factor. Nature 327: 524–526 doi:10.1038/327524a0 - DOI - PubMed
-
- Moncada S, Vane JR (1978) Pharmacology and endogenous roles of prostaglandin endoperoxides, thromboxane A2, and prostacyclin. Pharmacol Rev 30: 293–331. - PubMed
-
- Campbell WB, Gebremedhin D, Pratt PF, Harder DR (1996) Identification of epoxyeicosatrienoic acids as endothelium-derived hyperpolarizing factors. Circ Res 78: 415–423. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
