

An experimentally determined evolutionary model dramatically improves phylogenetic fit
- PMID: 24859245
- PMCID: PMC4104320
- DOI: 10.1093/molbev/msu173
An experimentally determined evolutionary model dramatically improves phylogenetic fit
Abstract
All modern approaches to molecular phylogenetics require a quantitative model for how genes evolve. Unfortunately, existing evolutionary models do not realistically represent the site-heterogeneous selection that governs actual sequence change. Attempts to remedy this problem have involved augmenting these models with a burgeoning number of free parameters. Here, I demonstrate an alternative: Experimental determination of a parameter-free evolutionary model via mutagenesis, functional selection, and deep sequencing. Using this strategy, I create an evolutionary model for influenza nucleoprotein that describes the gene phylogeny far better than existing models with dozens or even hundreds of free parameters. Emerging high-throughput experimental strategies such as the one employed here provide fundamentally new information that has the potential to transform the sensitivity of phylogenetic and genetic analyses.
Keywords: codon model; deep mutational scanning; influenza; nucleoprotein; phylogenetics; substitution model.
© The Author 2014. Published by Oxford University Press on behalf of the Society for Molecular Biology and Evolution.
Figures
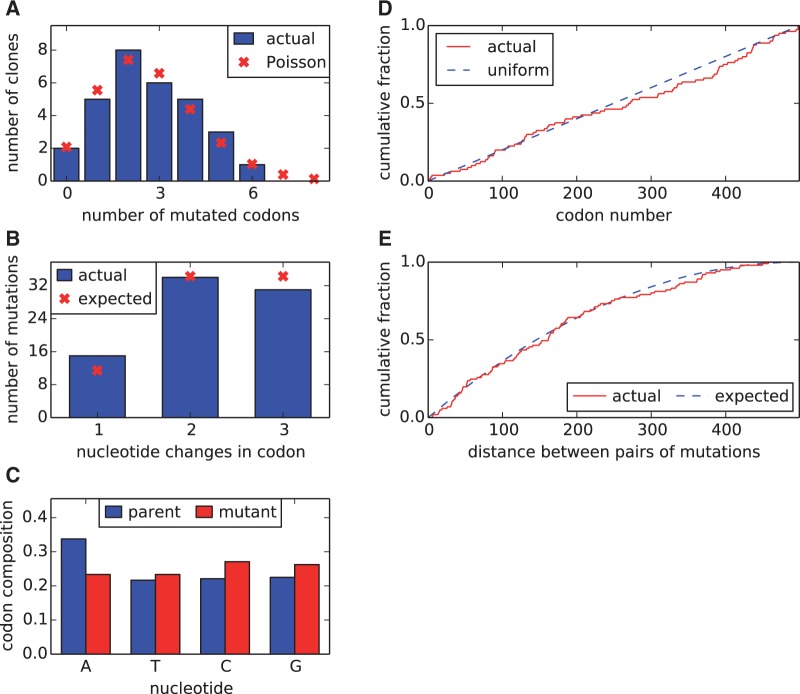

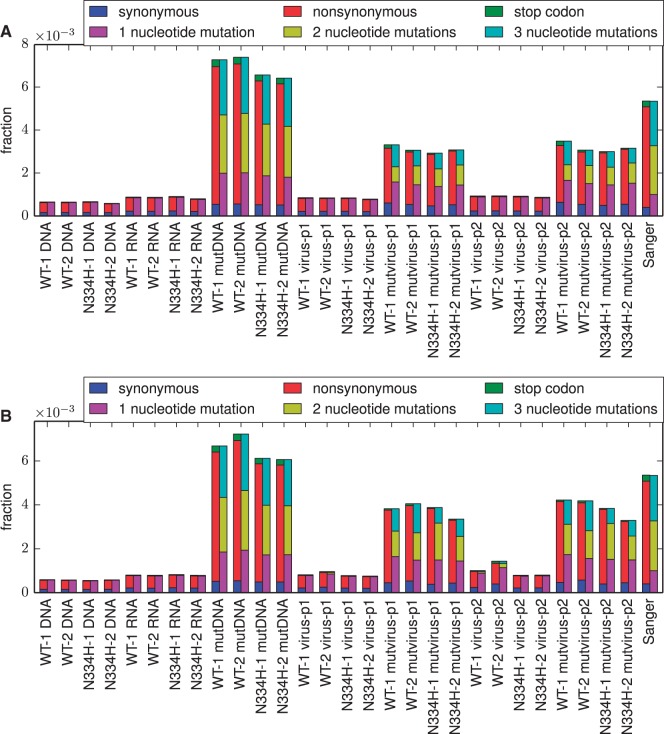
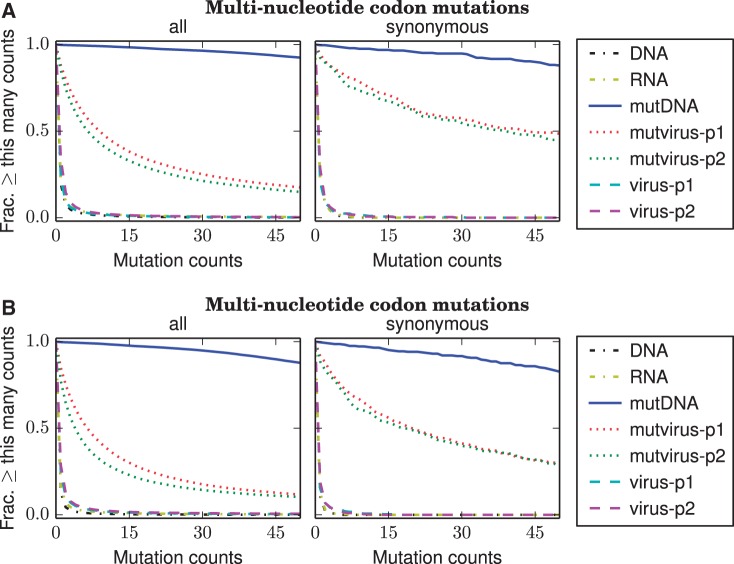


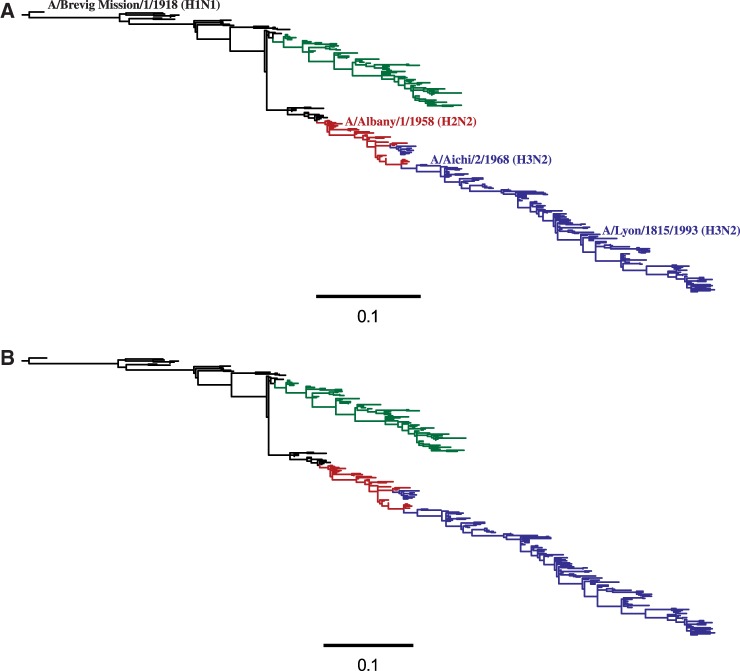
References
-
- Bershtein S, Segal M, Bekerman R, Tokuriki N, Tawfik DS. Robustness-epistasis link shapes the fitness landscape of a randomly drifting protein. Nature. 2006;444:929–932. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
