

Prions: generation and spread versus neurotoxicity
- PMID: 24860100
- PMCID: PMC4106307
- DOI: 10.1074/jbc.R114.568477
Prions: generation and spread versus neurotoxicity
Abstract
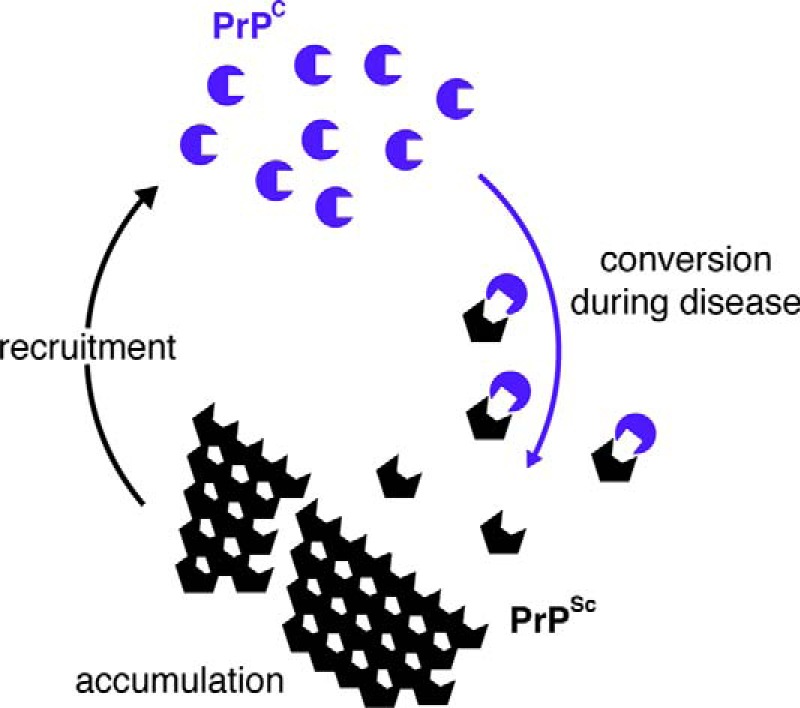
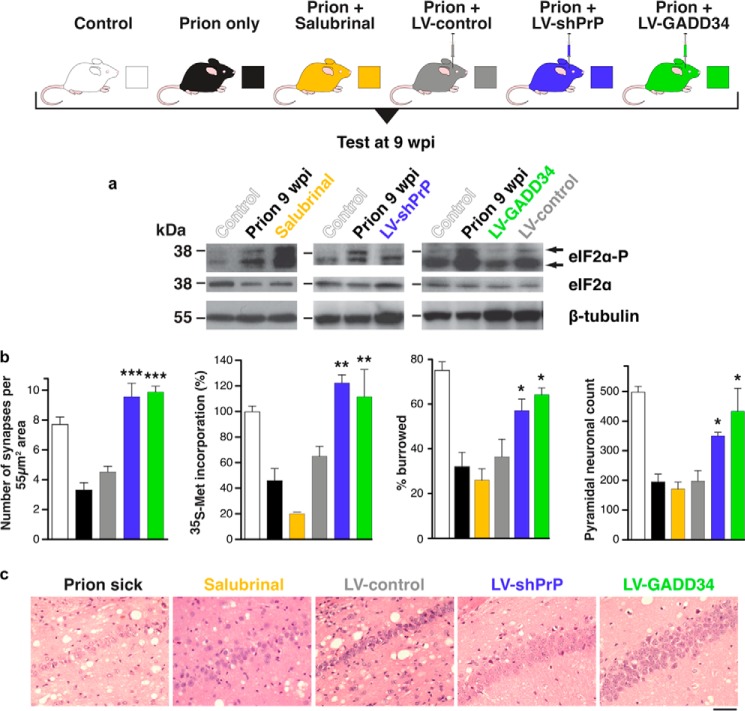
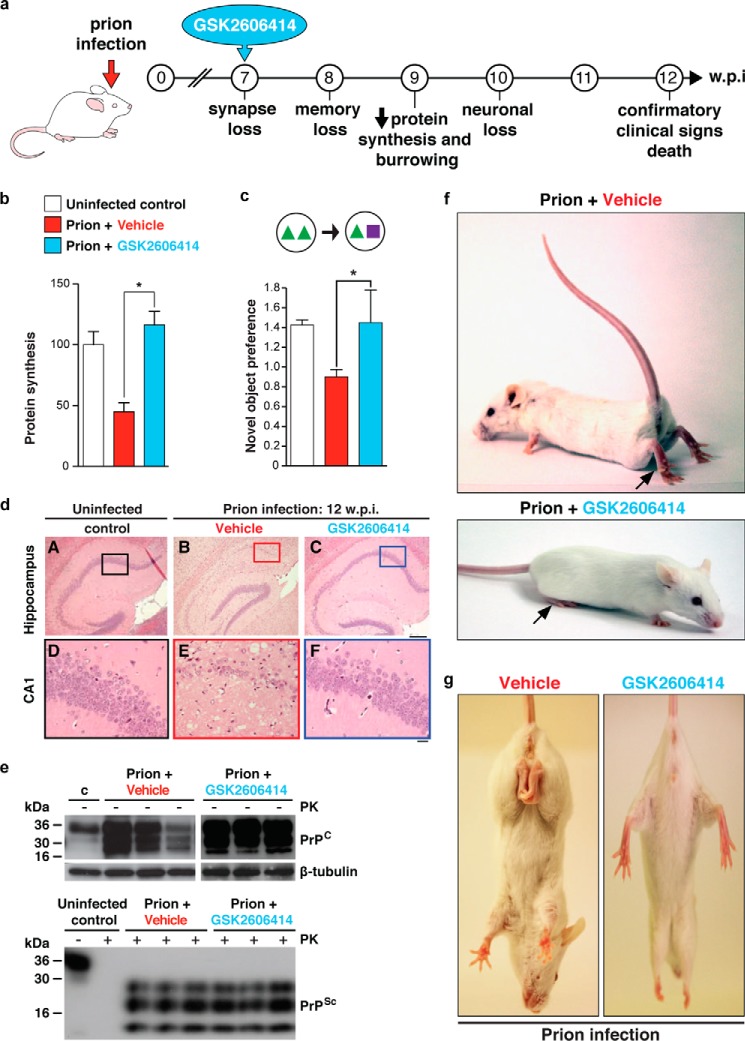
Neurodegenerative diseases are characterized by the aggregation of misfolded proteins in the brain. Among these disorders are the prion diseases, which are transmissible, and in which the misfolded proteins ("prions") are also the infectious agent. Increasingly, it appears that misfolded proteins in Alzheimer and Parkinson diseases and the tauopathies also propagate in a "prion-like" manner. However, the association between prion formation, spread, and neurotoxicity is not clear. Recently, we showed that in prion disease, protein misfolding leads to neurodegeneration through dysregulation of generic proteostatic mechanisms, specifically, the unfolded protein response. Genetic and pharmacological manipulation of the unfolded protein response was neuroprotective despite continuing prion replication, hence dissociating this from neurotoxicity. The data have clear implications for treatment across the spectrum of these disorders, targeting pathogenic processes downstream of protein misfolding.
Keywords: Alzheimer Disease; Alzheimer's; Gene Therapy; Neurodegeneration; Neuroprotection; Prion; Unfolded Protein Response (UPR).
© 2014 by The American Society for Biochemistry and Molecular Biology, Inc.
Figures

References
-
- Prusiner S. B. (1982) Novel proteinaceous infectious particles cause scrapie. Science 216, 136–144 - PubMed
-
- Griffith J. S. (1967) Self-replication and scrapie. Nature 215, 1043–1044 - PubMed
-
- McKinley M. P., Bolton D. C., Prusiner S. B. (1983) A protease-resistant protein is a structural component of the scrapie prion. Cell 35, 57–62 - PubMed
-
- Oesch B., Westaway D., Wälchli M., McKinley M. P., Kent S. B., Aebersold R., Barry R. A., Tempst P., Teplow D. B., Hood L. E., et al. (1985) A cellular gene encodes scrapie PrP 27–30 protein. Cell 40, 735–746 - PubMed
-
- Chesebro B., Race R., Wehrly K., Nishio J., Bloom M., Lechner D., Bergstrom S., Robbins K., Mayer L., Keith J. M., et al. (1985) Identification of scrapie prion protein-specific mRNA in scrapie-infected and uninfected brain. Nature 315, 331–333 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
