

Mitochondrial dysfunction induced by frataxin deficiency is associated with cellular senescence and abnormal calcium metabolism
- PMID: 24860428
- PMCID: PMC4026758
- DOI: 10.3389/fncel.2014.00124
Mitochondrial dysfunction induced by frataxin deficiency is associated with cellular senescence and abnormal calcium metabolism
Abstract
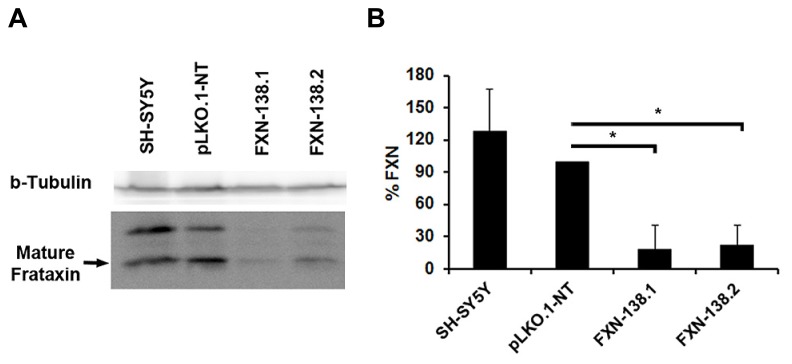
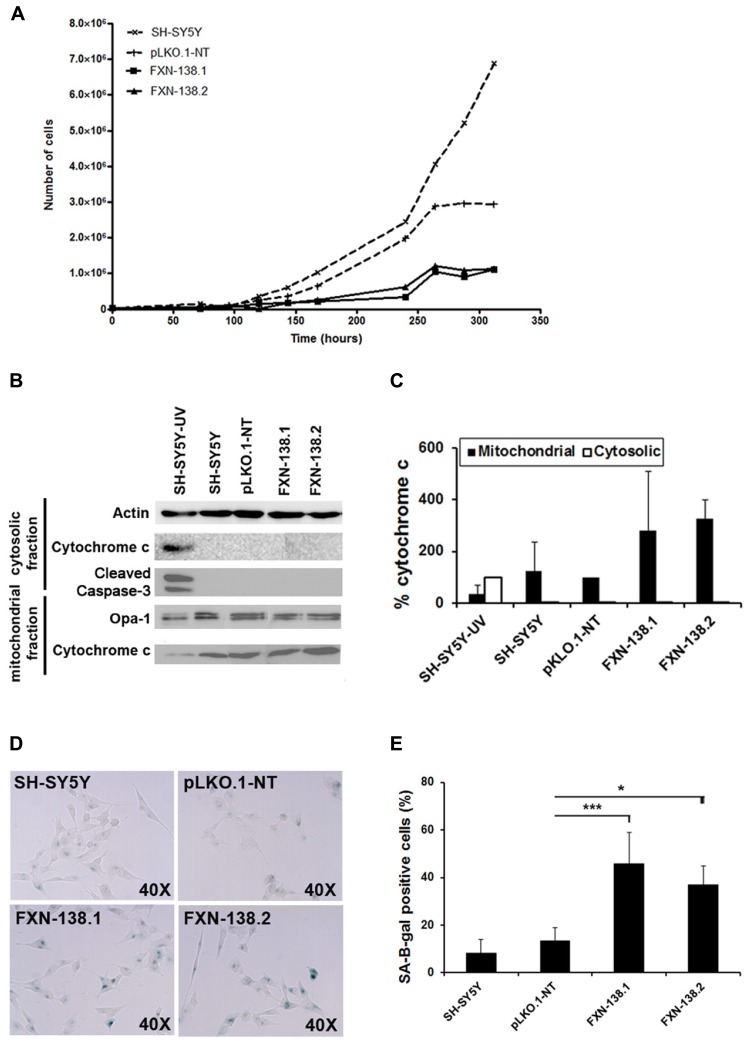
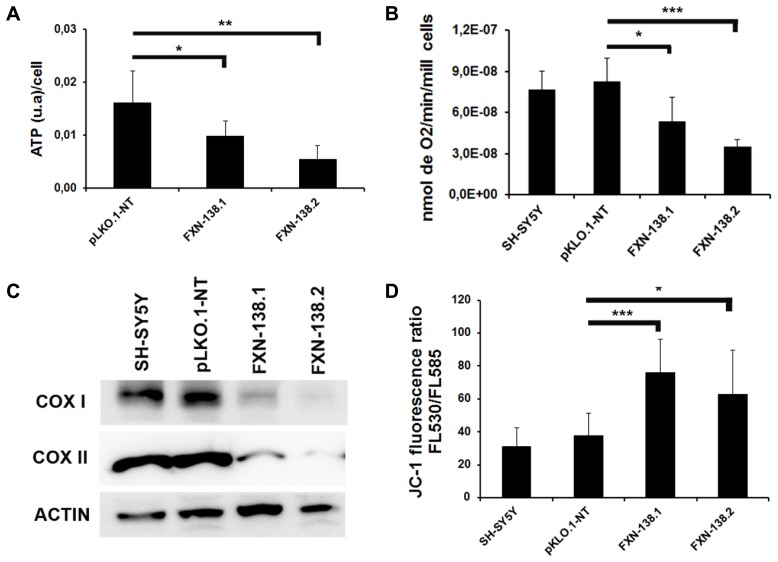
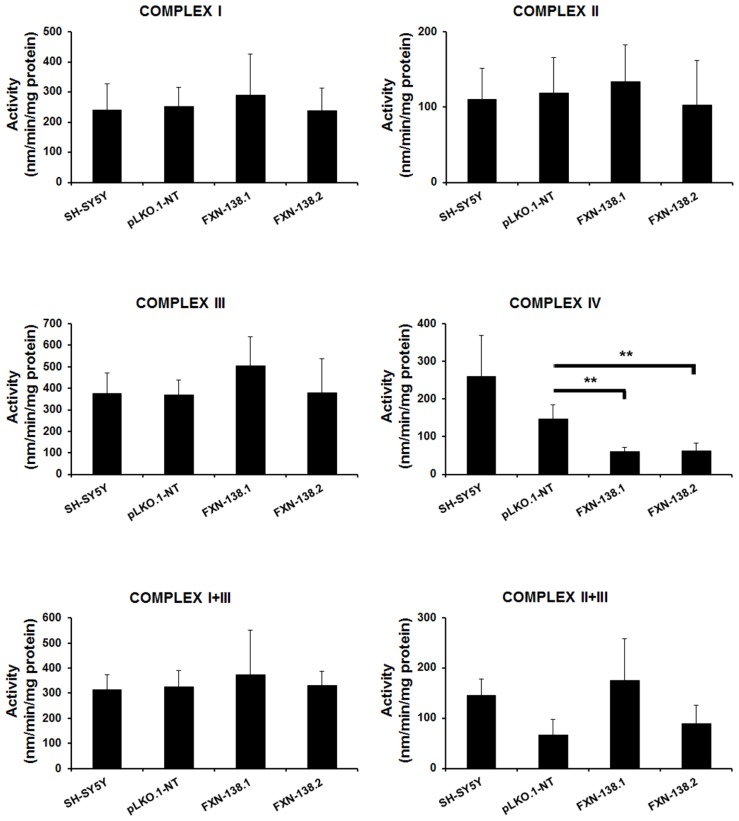
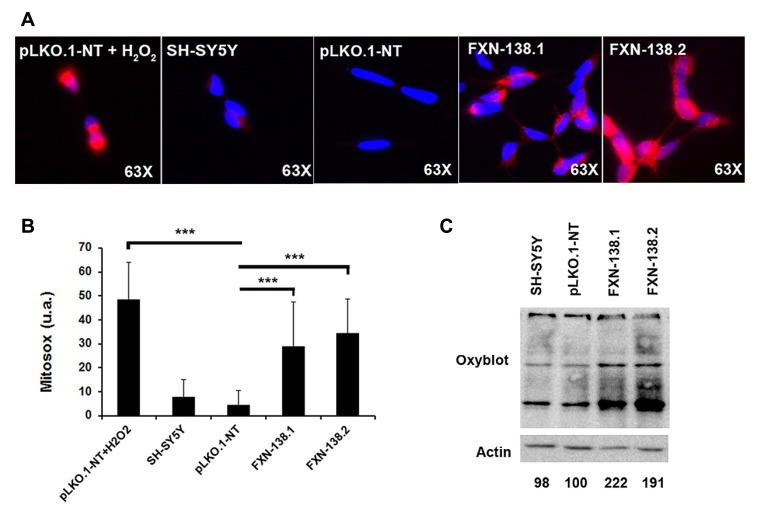
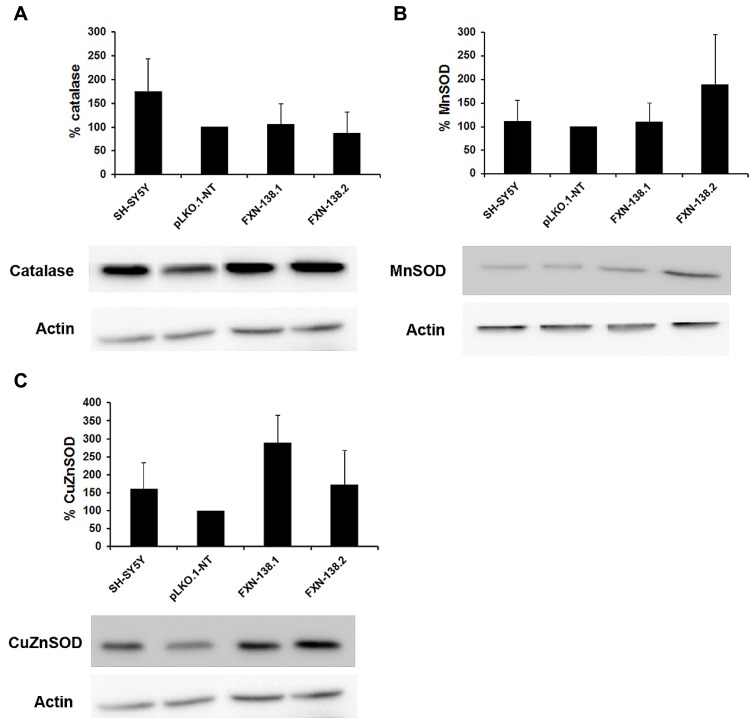
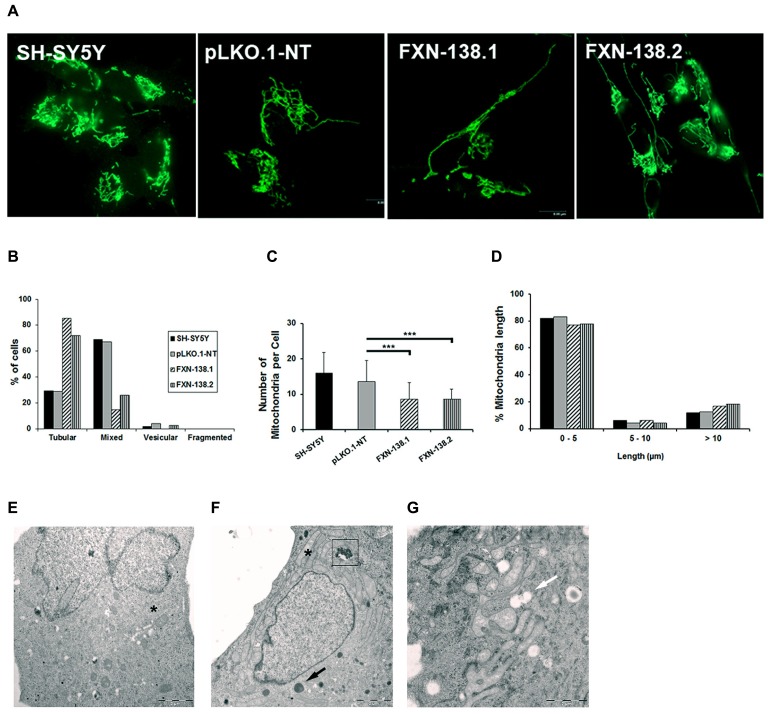
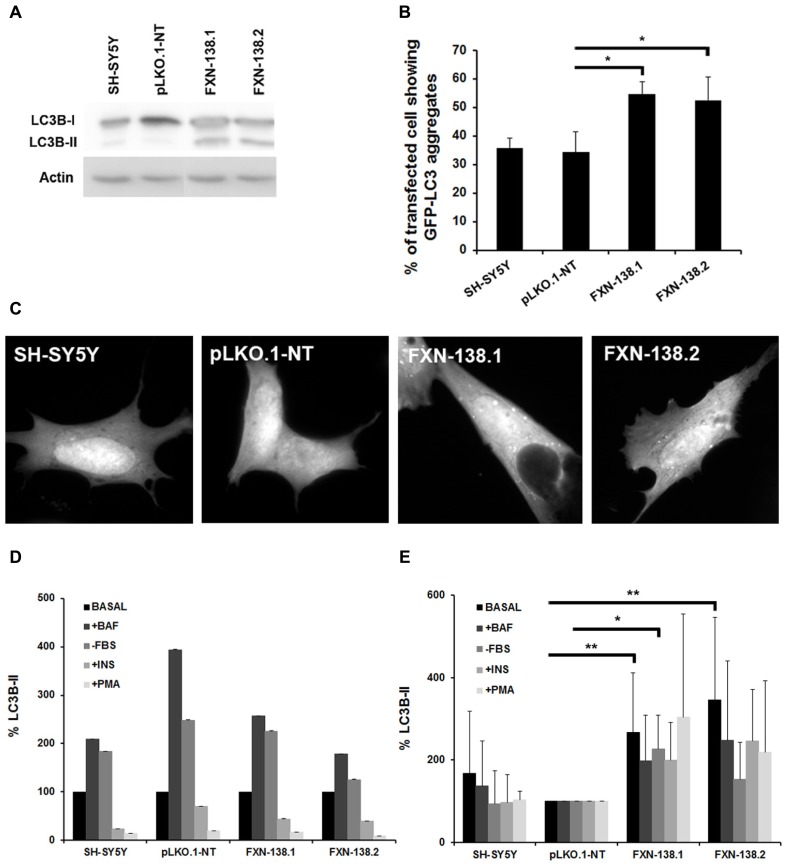
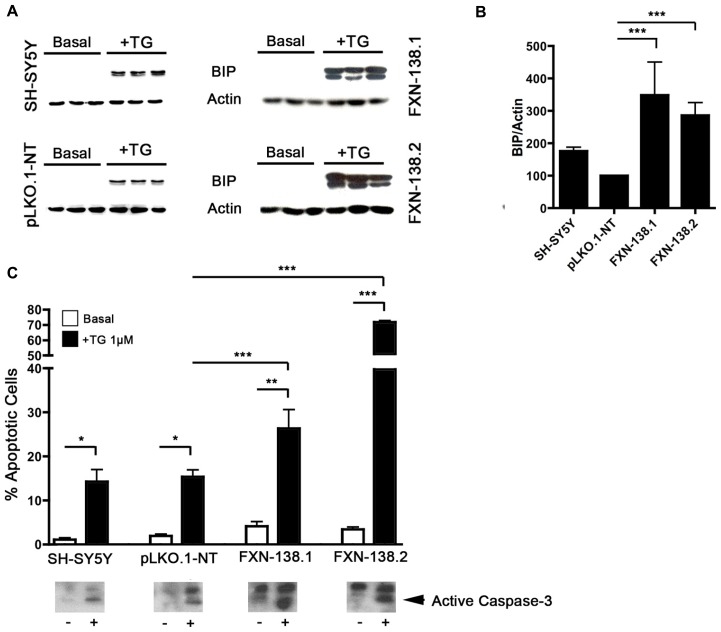
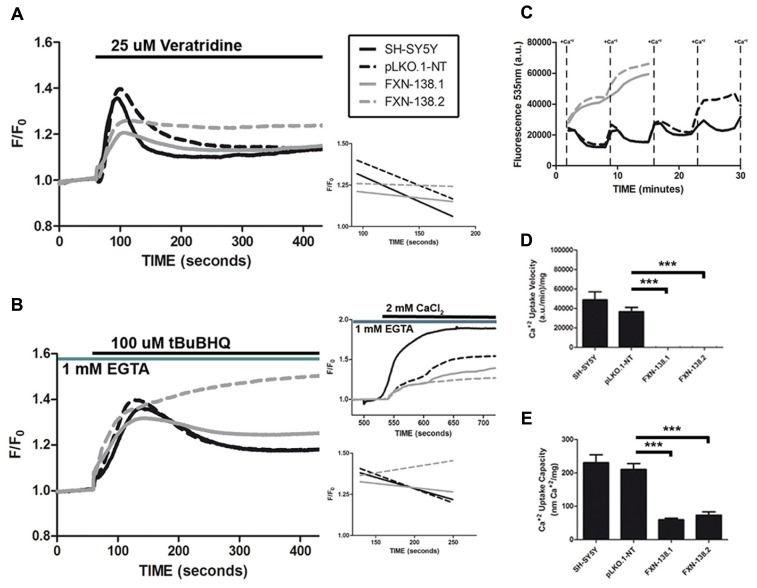
Friedreich ataxia is considered a neurodegenerative disorder involving both the peripheral and central nervous systems. Dorsal root ganglia (DRG) are the major target tissue structures. This neuropathy is caused by mutations in the FXN gene that encodes frataxin. Here, we investigated the mitochondrial and cell consequences of frataxin depletion in a cellular model based on frataxin silencing in SH-SY5Y human neuroblastoma cells, a cell line that has been used widely as in vitro models for studies on neurological diseases. We showed that the reduction of frataxin induced mitochondrial dysfunction due to a bioenergetic deficit and abnormal Ca(2+) homeostasis in the mitochondria that were associated with oxidative and endoplasmic reticulum stresses. The depletion of frataxin did not cause cell death but increased autophagy, which may have a cytoprotective effect against cellular insults such as oxidative stress. Frataxin silencing provoked slow cell growth associated with cellular senescence, as demonstrated by increased SA-βgal activity and cell cycle arrest at the G1 phase. We postulate that cellular senescence might be related to a hypoplastic defect in the DRG during neurodevelopment, as suggested by necropsy studies.
Keywords: ER-stress; Friedreich ataxia; autophagy; calcium metabolism; cellular senescence; frataxin; mitochondrial dysfunction.
Figures
References
-
- Al-Mahdawi S., Pinto R. M., Varshney D., Lawrence L., Lowrie M. B., Hughes S., et al. (2006). GAA repeat expansion mutation mouse models of Friedreich ataxia exhibit oxidative stress leading to progressive neuronal and cardiac pathology. Genomics 88 580–590 10.1016/j.ygeno.2006.06.015 - DOI - PMC - PubMed
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous