

Exploring genome wide bisulfite sequencing for DNA methylation analysis in livestock: a technical assessment
- PMID: 24860595
- PMCID: PMC4026711
- DOI: 10.3389/fgene.2014.00126
Exploring genome wide bisulfite sequencing for DNA methylation analysis in livestock: a technical assessment
Abstract
Recent advances made in "omics" technologies are contributing to a revolution in livestock selection and breeding practices. Epigenetic mechanisms, including DNA methylation are important determinants for the control of gene expression in mammals. DNA methylation research will help our understanding of how environmental factors contribute to phenotypic variation of complex production and health traits. High-throughput sequencing is a vital tool for the comprehensive analysis of DNA methylation, and bisulfite-based strategies coupled with DNA sequencing allows for quantitative, site-specific methylation analysis at the genome level or genome wide. Reduced representation bisulfite sequencing (RRBS) and more recently whole genome bisulfite sequencing (WGBS) have proven to be effective techniques for studying DNA methylation in both humans and mice. Here we report the development of RRBS and WGBS for use in sheep, the first application of this technology in livestock species. Important technical issues associated with these methodologies including fragment size selection and sequence depth are examined and discussed.
Keywords: DNA methylation; RRBS; WGBS; epigenetics; fragment size; quantification; sheep.
Figures
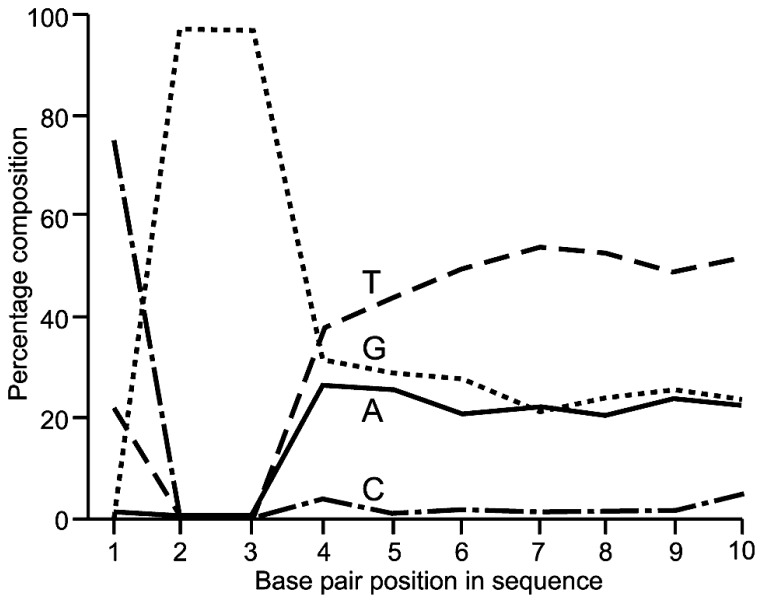
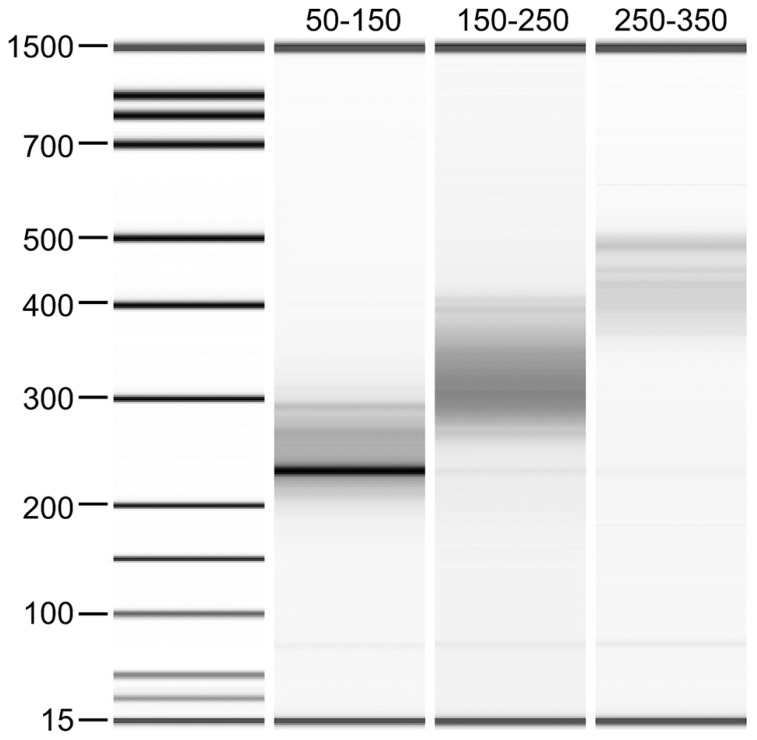
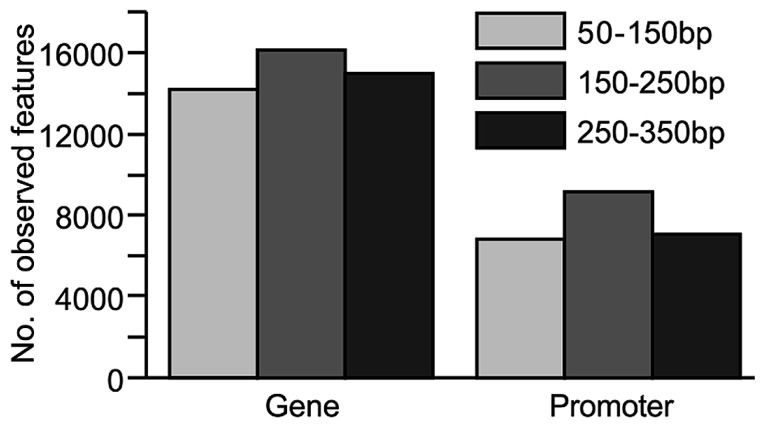
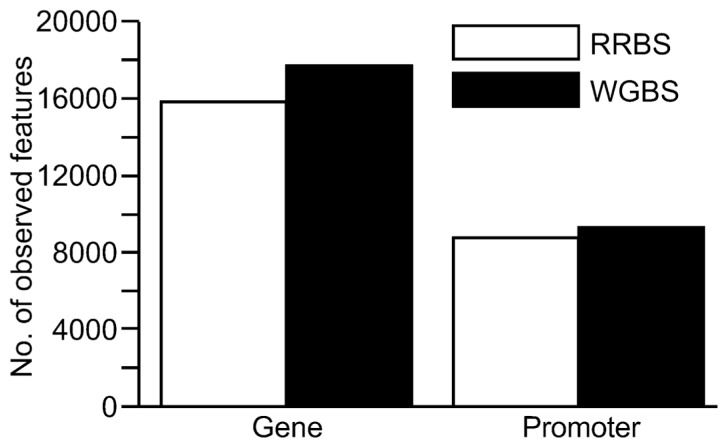
Similar articles
-
Plant-RRBS, a bisulfite and next-generation sequencing-based methylome profiling method enriching for coverage of cytosine positions.BMC Plant Biol. 2017 Jul 6;17(1):115. doi: 10.1186/s12870-017-1070-y. BMC Plant Biol. 2017. PMID: 28683715 Free PMC article.
-
DNA methylation estimation using methylation-sensitive restriction enzyme bisulfite sequencing (MREBS).PLoS One. 2019 Apr 4;14(4):e0214368. doi: 10.1371/journal.pone.0214368. eCollection 2019. PLoS One. 2019. PMID: 30946758 Free PMC article.
-
Studying DNA Methylation Genome-Wide by Bisulfite Sequencing from Low Amounts of DNA in Mammals.Methods Mol Biol. 2021;2214:207-220. doi: 10.1007/978-1-0716-0958-3_14. Methods Mol Biol. 2021. PMID: 32944912
-
Global DNA methylation profiling technologies and the ovarian cancer methylome.Methods Mol Biol. 2015;1238:653-75. doi: 10.1007/978-1-4939-1804-1_34. Methods Mol Biol. 2015. PMID: 25421685 Review.
-
[The application of next generation sequencing on epigenetic study].Yi Chuan. 2014 Mar;36(3):256-75. Yi Chuan. 2014. PMID: 24846966 Review. Chinese.
Cited by
-
Genetic Determinism Exists for the Global DNA Methylation Rate in Sheep.Front Genet. 2020 Dec 23;11:616960. doi: 10.3389/fgene.2020.616960. eCollection 2020. Front Genet. 2020. PMID: 33424937 Free PMC article.
-
Decoding the transcriptomic expression and genomic methylation patterns in the tendon proper and its peritenon region in the aging horse.BMC Res Notes. 2023 Oct 11;16(1):267. doi: 10.1186/s13104-023-06562-1. BMC Res Notes. 2023. PMID: 37821884 Free PMC article.
-
Tumour Heterogeneity: The Key Advantages of Single-Cell Analysis.Int J Mol Sci. 2016 Dec 20;17(12):2142. doi: 10.3390/ijms17122142. Int J Mol Sci. 2016. PMID: 27999407 Free PMC article. Review.
-
4mCPred-CNN-Prediction of DNA N4-Methylcytosine in the Mouse Genome Using a Convolutional Neural Network.Genes (Basel). 2021 Feb 20;12(2):296. doi: 10.3390/genes12020296. Genes (Basel). 2021. PMID: 33672576 Free PMC article.
-
Rapid Multiplexed Reduced Representation Bisulfite Sequencing Library Prep (rRRBS).Bio Protoc. 2019 Feb 20;9(4):e3171. doi: 10.21769/BioProtoc.3171. eCollection 2019 Feb 20. Bio Protoc. 2019. PMID: 33654977 Free PMC article.
References
LinkOut - more resources
Full Text Sources
Other Literature Sources