

B lymphocytes undergo TLR2-dependent apoptosis upon Shigella infection
- PMID: 24863068
- PMCID: PMC4042640
- DOI: 10.1084/jem.20130914
B lymphocytes undergo TLR2-dependent apoptosis upon Shigella infection
Abstract
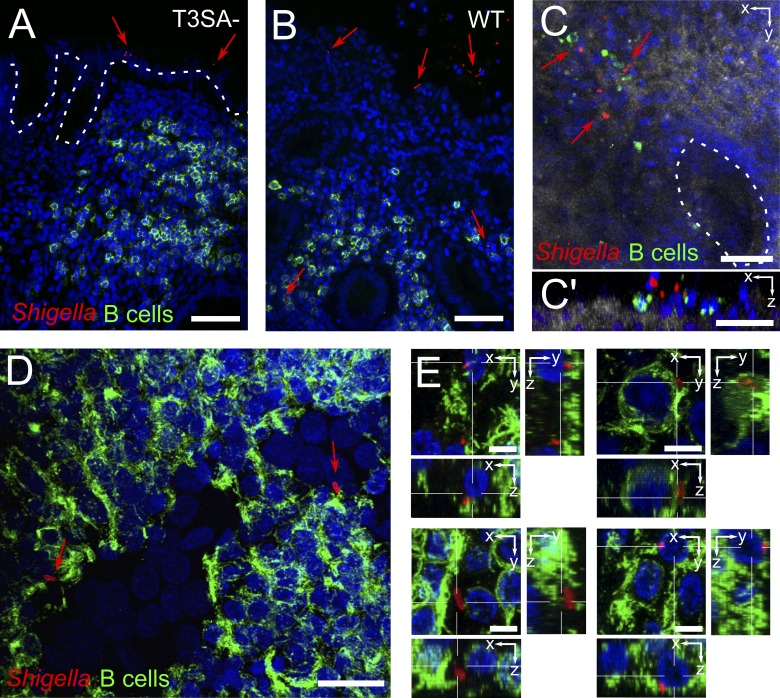
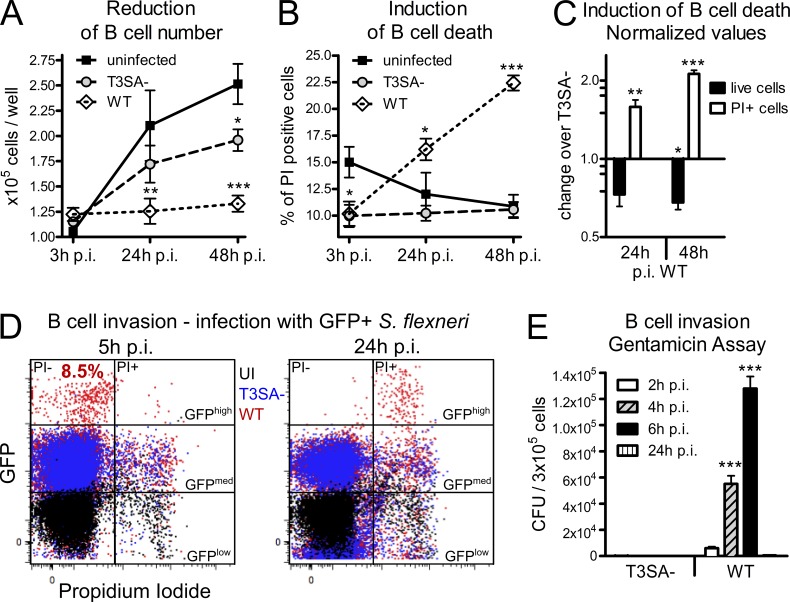
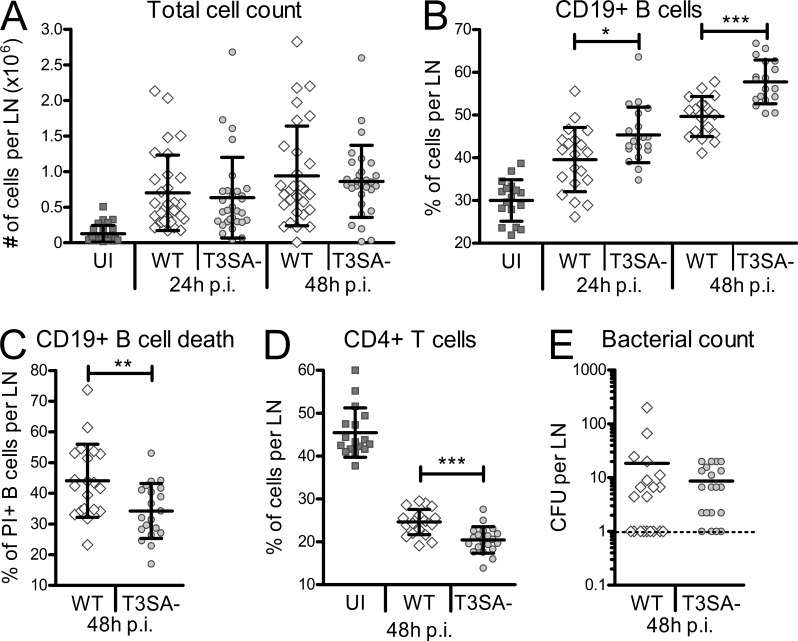
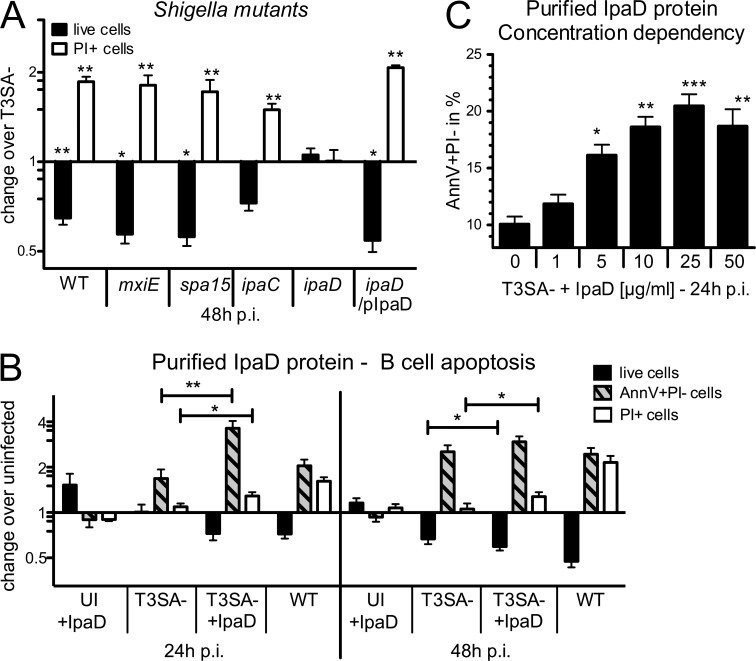
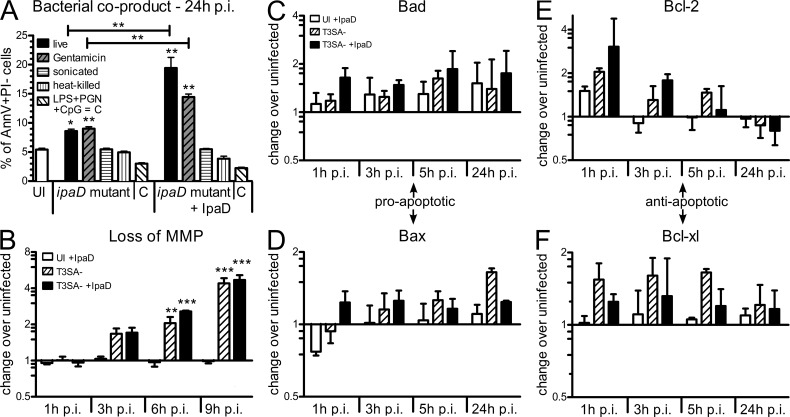
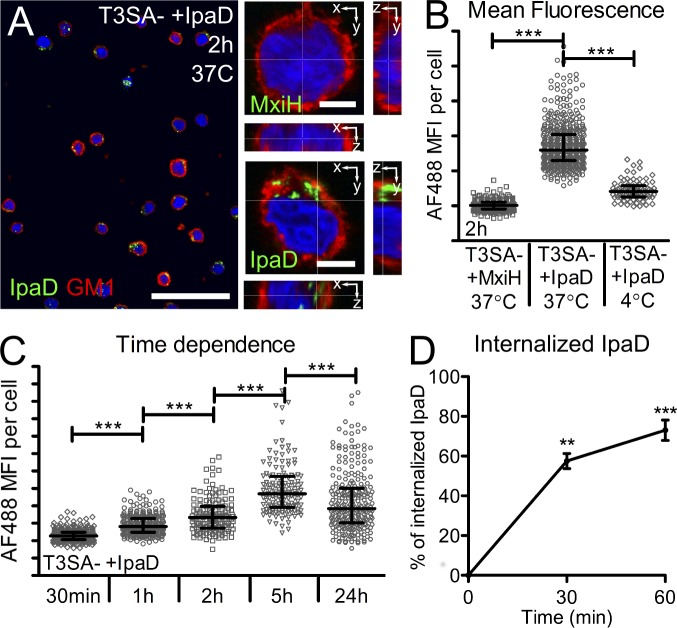
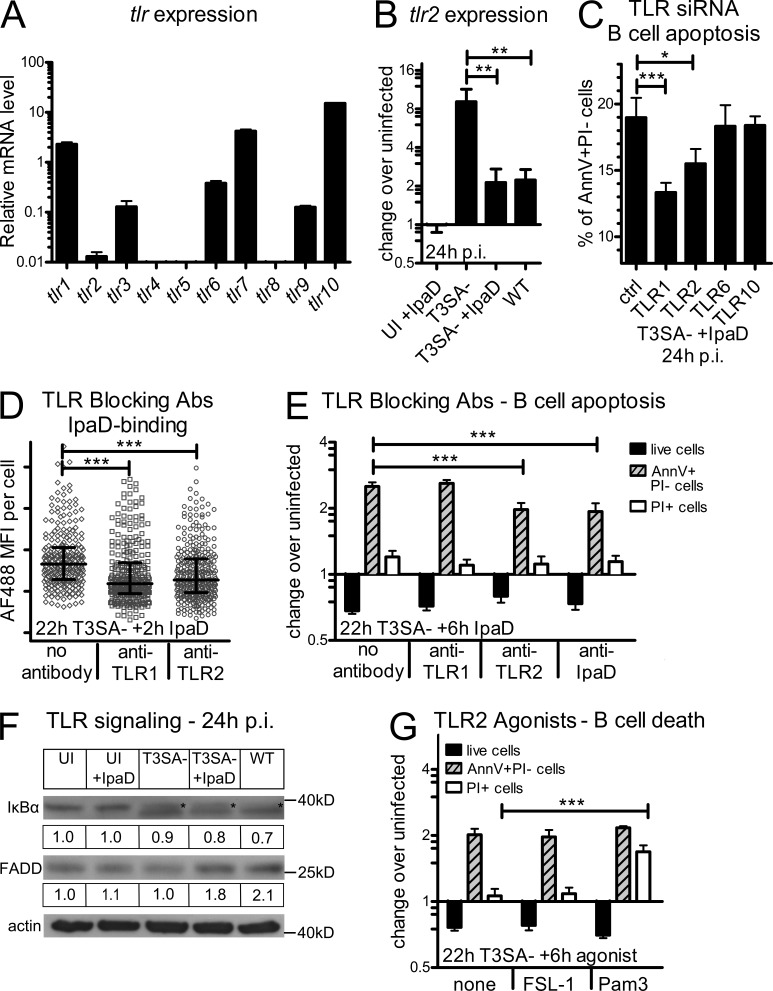
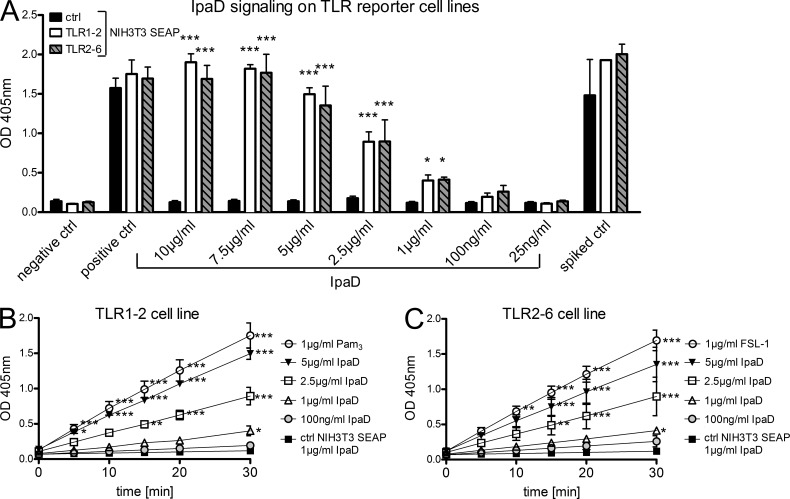
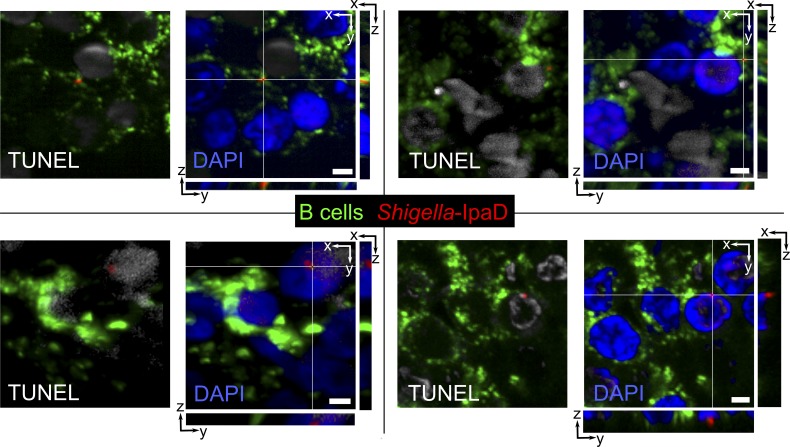
Antibody-mediated immunity to Shigella, the causative agent of bacillary dysentery, requires several episodes of infection to get primed and is short-lasting, suggesting that the B cell response is functionally impaired. We show that upon ex vivo infection of human colonic tissue, invasive S. flexneri interacts with and occasionally invades B lymphocytes. The induction of a type three secretion apparatus (T3SA)-dependent B cell death is observed in the human CL-01 B cell line in vitro, as well as in mouse B lymphocytes in vivo. In addition to cell death occurring in Shigella-invaded CL-01 B lymphocytes, we provide evidence that the T3SA needle tip protein IpaD can induce cell death in noninvaded cells. IpaD binds to and induces B cell apoptosis via TLR2, a signaling receptor thus far considered to result in activation of B lymphocytes. The presence of bacterial co-signals is required to sensitize B cells to apoptosis and to up-regulate tlr2, thus enhancing IpaD binding. Apoptotic B lymphocytes in contact with Shigella-IpaD are detected in rectal biopsies of infected individuals. This study therefore adds direct B lymphocyte targeting to the diversity of mechanisms used by Shigella to dampen the host immune response.
© 2014 Nothelfer et al.
Figures
Similar articles
-
Macrophage Apoptosis Triggered by IpaD from Shigella flexneri.Infect Immun. 2016 May 24;84(6):1857-1865. doi: 10.1128/IAI.01483-15. Print 2016 Jun. Infect Immun. 2016. PMID: 27068089 Free PMC article.
-
Conformational changes in IpaD from Shigella flexneri upon binding bile salts provide insight into the second step of type III secretion.Biochemistry. 2011 Jan 18;50(2):172-80. doi: 10.1021/bi101365f. Epub 2010 Dec 15. Biochemistry. 2011. PMID: 21126091 Free PMC article.
-
Vaccination With Mouse Dendritic Cells Loaded With an IpaD-IpaB Fusion Provides Protection Against Shigellosis.Front Immunol. 2019 Feb 8;10:192. doi: 10.3389/fimmu.2019.00192. eCollection 2019. Front Immunol. 2019. PMID: 30800131 Free PMC article.
-
Outer membrane protein A (OmpA) from Shigella flexneri 2a: a promising subunit vaccine candidate.Vaccine. 2013 Aug 12;31(36):3644-50. doi: 10.1016/j.vaccine.2013.05.100. Epub 2013 Jun 10. Vaccine. 2013. PMID: 23764536 Review.
-
Implications of Spatiotemporal Regulation of Shigella flexneri Type Three Secretion Activity on Effector Functions: Think Globally, Act Locally.Front Cell Infect Microbiol. 2016 Mar 9;6:28. doi: 10.3389/fcimb.2016.00028. eCollection 2016. Front Cell Infect Microbiol. 2016. PMID: 27014638 Free PMC article. Review.
Cited by
-
Pathogen manipulation of B cells: the best defence is a good offence.Nat Rev Microbiol. 2015 Mar;13(3):173-84. doi: 10.1038/nrmicro3415. Epub 2015 Feb 9. Nat Rev Microbiol. 2015. PMID: 25659322 Review.
-
The Intriguing Interaction of Escherichia coli with the Host Environment and Innovative Strategies To Interfere with Colonization: a Summary of the 2019 E. coli and the Mucosal Immune System Meeting.Appl Environ Microbiol. 2020 Nov 24;86(24):e02085-20. doi: 10.1128/AEM.02085-20. Print 2020 Nov 24. Appl Environ Microbiol. 2020. PMID: 33008822 Free PMC article.
-
Production of egg yolk antibody (IgY) against a chimeric protein containing IpaD, StxB, and TolC antigens from Shigella: An investigation of its prophylactic effects against Shiga toxin (Stx) and Shigella dysenteriae in vitro and in vivo.Heliyon. 2024 Feb 16;10(4):e26361. doi: 10.1016/j.heliyon.2024.e26361. eCollection 2024 Feb 29. Heliyon. 2024. PMID: 38404796 Free PMC article.
-
Trial Watch: Immunostimulation with Toll-like receptor agonists in cancer therapy.Oncoimmunology. 2015 Sep 2;5(3):e1088631. doi: 10.1080/2162402X.2015.1088631. eCollection 2016 Mar. Oncoimmunology. 2015. PMID: 27141345 Free PMC article. Review.
-
HrpE, the major component of the Xanthomonas type three protein secretion pilus, elicits plant immunity responses.Sci Rep. 2018 Jun 29;8(1):9842. doi: 10.1038/s41598-018-27869-1. Sci Rep. 2018. PMID: 29959345 Free PMC article.
References
-
- Acosta Rodriguez E.V., Zuniga E.I., Montes C.L., Merino M.C., Bermejo D.A., Amezcua Vesely M.C., Motran C.C., Gruppi A. 2007. Trypanosoma cruzi infection beats the B-cell compartment favouring parasite establishment: can we strike first? Scand. J. Immunol. 66:137–142 10.1111/j.1365-3083.2007.01968.x - DOI - PubMed
-
- Ahmed F., Clemens J.D., Rao M.R., Sack D.A., Khan M.R., Haque E. 1992. Community-based evaluation of the effect of breast-feeding on the risk of microbiologically confirmed or clinically presumptive shigellosis in Bangladeshi children. Pediatrics. 90:406–411 - PubMed
-
- Bhunia A.K., Feng X. 1999. Examination of cytopathic effect and apoptosis in Listeria monocytogenes-infected hybridoma B-lymphocyte (Ped-2E9) line in vitro. J. Microbiol. Biotechnol. 9:398–403
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
