

At the Bench: Helicobacter pylori, dysregulated host responses, DNA damage, and gastric cancer
- PMID: 24868089
- PMCID: PMC4101087
- DOI: 10.1189/jlb.4BT0214-099R
At the Bench: Helicobacter pylori, dysregulated host responses, DNA damage, and gastric cancer
Abstract
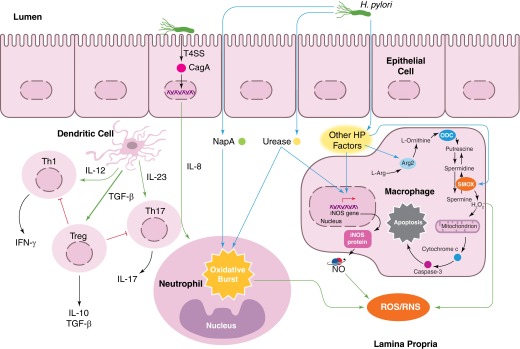
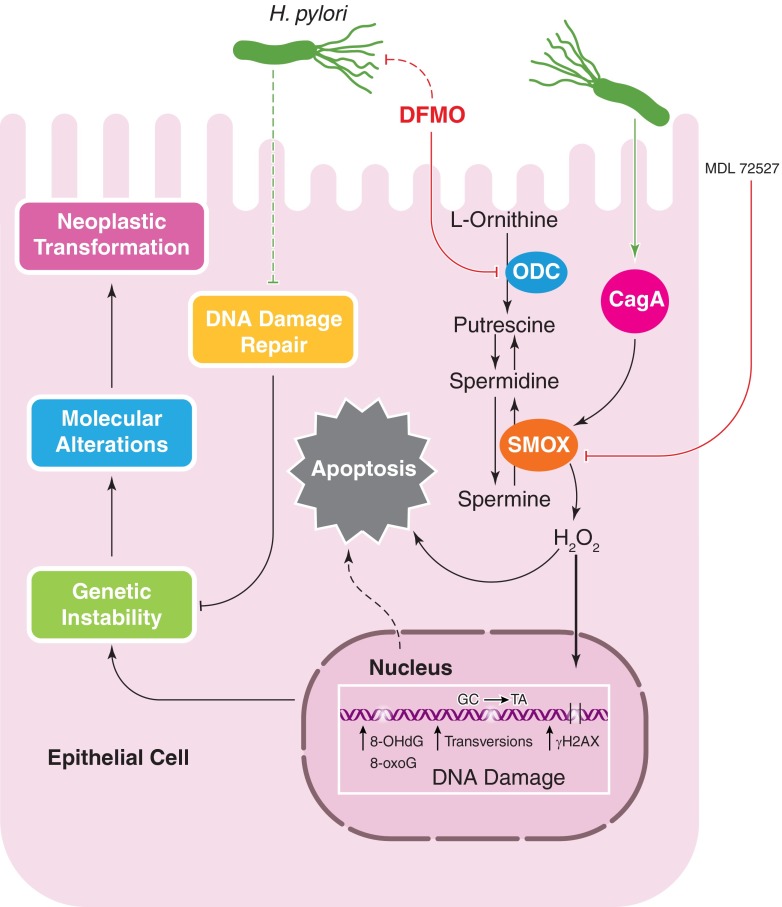
Helicobacter pylori infection is the strongest known risk factor for the development of gastric cancer. Given that ∼50% of the global population is infected with this pathogen, there is great impetus to elucidate underlying causes that mediate progression from infection to cancer. Recent evidence suggests that H. pylori-induced chronic inflammation and oxidative stress create an environment conducive to DNA damage and tissue injury. DNA damage leads to genetic instability and eventually, neoplastic transformation. Pathogen-encoded virulence factors induce a robust but futile immune response and alter host pathways that lower the threshold for carcinogenesis, including DNA damage repair, polyamine synthesis and catabolism, antioxidant responses, and cytokine production. Collectively, such dysregulation creates a protumorigenic microenvironment within the stomach. This review seeks to address each of these aspects of H. pylori infection and to call attention to areas of particular interest within this field of research. This review also seeks to prioritize areas of translational research related to H. pylori-induced gastric cancer based on insights garnered from basic research in this field. See related review by Dalal and Moss, At the Bedside: H. pylori, dysregulated host responses, DNA damage, and gastric cancer.
Keywords: DFMO; carcinogenesis; oxidative stress.
© 2014 Society for Leukocyte Biology.
Figures
References
-
- Parsonnet J., Friedman G. D., Vandersteen D. P., Chang Y., Vogelman J. H., Orentreich N., Sibley R. K. (1991) Helicobacter pylori infection and the risk of gastric carcinoma. N. Engl. J. Med. 325, 1127–1131 - PubMed
-
- Nomura A., Stemmermann G., Chyou P., Kato I., Perez-Perez G., Blaser M. (1991) Helicobacter pylori infection and gastric carcinoma among Japanese Americans in Hawaii. N. Engl. J. Med. 325, 1132–1136 - PubMed
-
- Peek R. M., Blaser M. J. (2002) Helicobacter pylori and gastrointestinal tract adenocarcinomas. Nat. Rev. Cancer 2, 28–37 - PubMed
-
- De Sablet T., Piazuelo M. B., Shaffer C. L., Schneider B. G., Asim M., Chaturvedi R., Bravo L. E., Sicinschi L. A., Delgado A. G., Mera R. M., Israel D. A., Romero-Gallo J., Peek R. M., Cover T. L., Correa P., Wilson K. T. (2011) Phylogeographic origin of Helicobacter pylori is a determinant of gastric cancer risk. Gut 60, 1189–1195 - PMC - PubMed
Publication types
MeSH terms
Grants and funding
- P01CA116087/CA/NCI NIH HHS/United States
- R01CA77955/CA/NCI NIH HHS/United States
- P30 DK058404/DK/NIDDK NIH HHS/United States
- R01 AT004821/AT/NCCIH NIH HHS/United States
- 5T32GM008554/GM/NIGMS NIH HHS/United States
- P01 CA116087/CA/NCI NIH HHS/United States
- T32 GM008554/GM/NIGMS NIH HHS/United States
- P30DK058404/DK/NIDDK NIH HHS/United States
- UL1 RR024975/RR/NCRR NIH HHS/United States
- R01 DK058587/DK/NIDDK NIH HHS/United States
- R01DK58587/DK/NIDDK NIH HHS/United States
- R01DK053620/DK/NIDDK NIH HHS/United States
- P01CA028842/CA/NCI NIH HHS/United States
- R01 DK053620/DK/NIDDK NIH HHS/United States
- P01 CA028842/CA/NCI NIH HHS/United States
- R01 CA077955/CA/NCI NIH HHS/United States
- R01AT004821/AT/NCCIH NIH HHS/United States
- UL1RR024975/RR/NCRR NIH HHS/United States
- I01 BX001453/BX/BLRD VA/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
