

Resistance mechanisms for the Bruton's tyrosine kinase inhibitor ibrutinib
- PMID: 24869598
- PMCID: PMC4144824
- DOI: 10.1056/NEJMoa1400029
Resistance mechanisms for the Bruton's tyrosine kinase inhibitor ibrutinib
Abstract
Background: Ibrutinib is an irreversible inhibitor of Bruton's tyrosine kinase (BTK) and is effective in chronic lymphocytic leukemia (CLL). Resistance to irreversible kinase inhibitors and resistance associated with BTK inhibition have not been characterized. Although only a small proportion of patients have had a relapse during ibrutinib therapy, an understanding of resistance mechanisms is important. We evaluated patients with relapsed disease to identify mutations that may mediate ibrutinib resistance.
Methods: We performed whole-exome sequencing at baseline and the time of relapse on samples from six patients with acquired resistance to ibrutinib therapy. We then performed functional analysis of identified mutations. In addition, we performed Ion Torrent sequencing for identified resistance mutations on samples from nine patients with prolonged lymphocytosis.
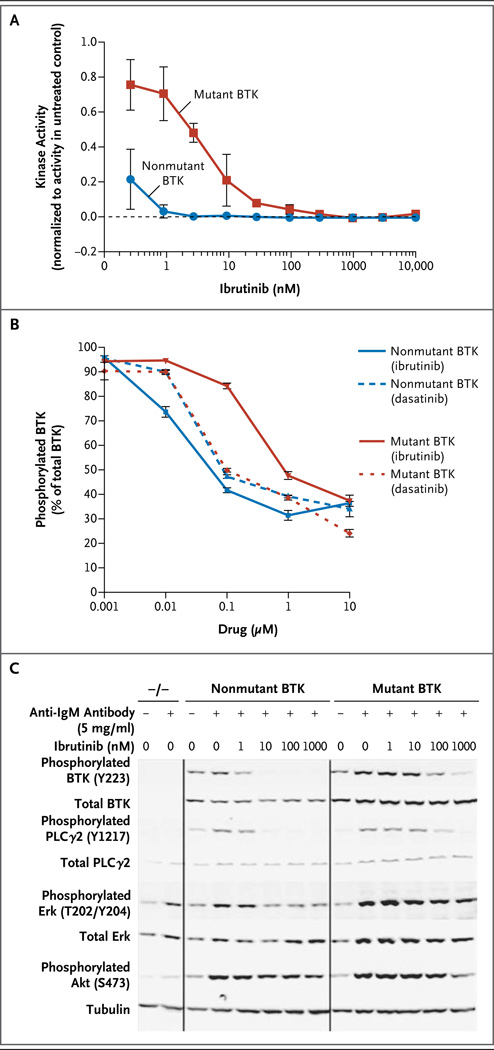
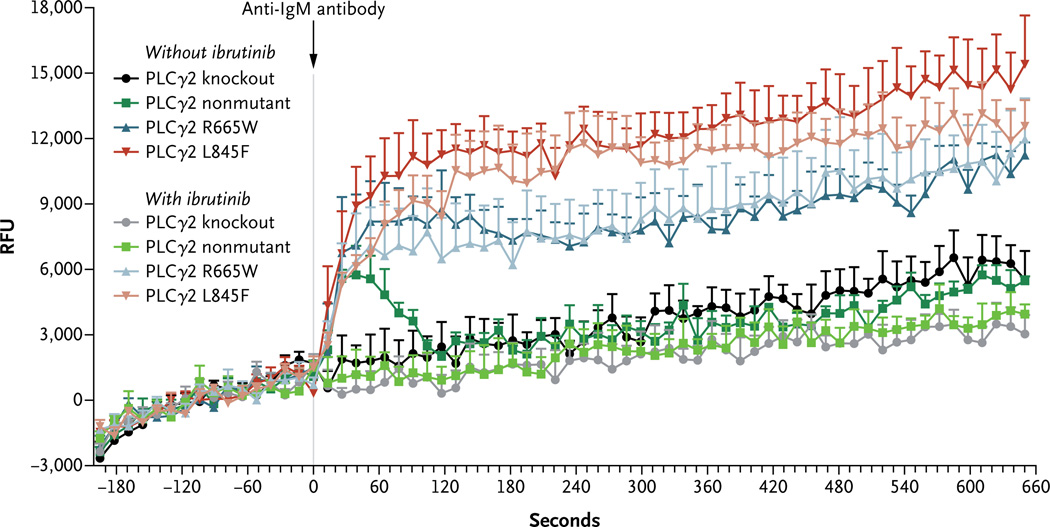
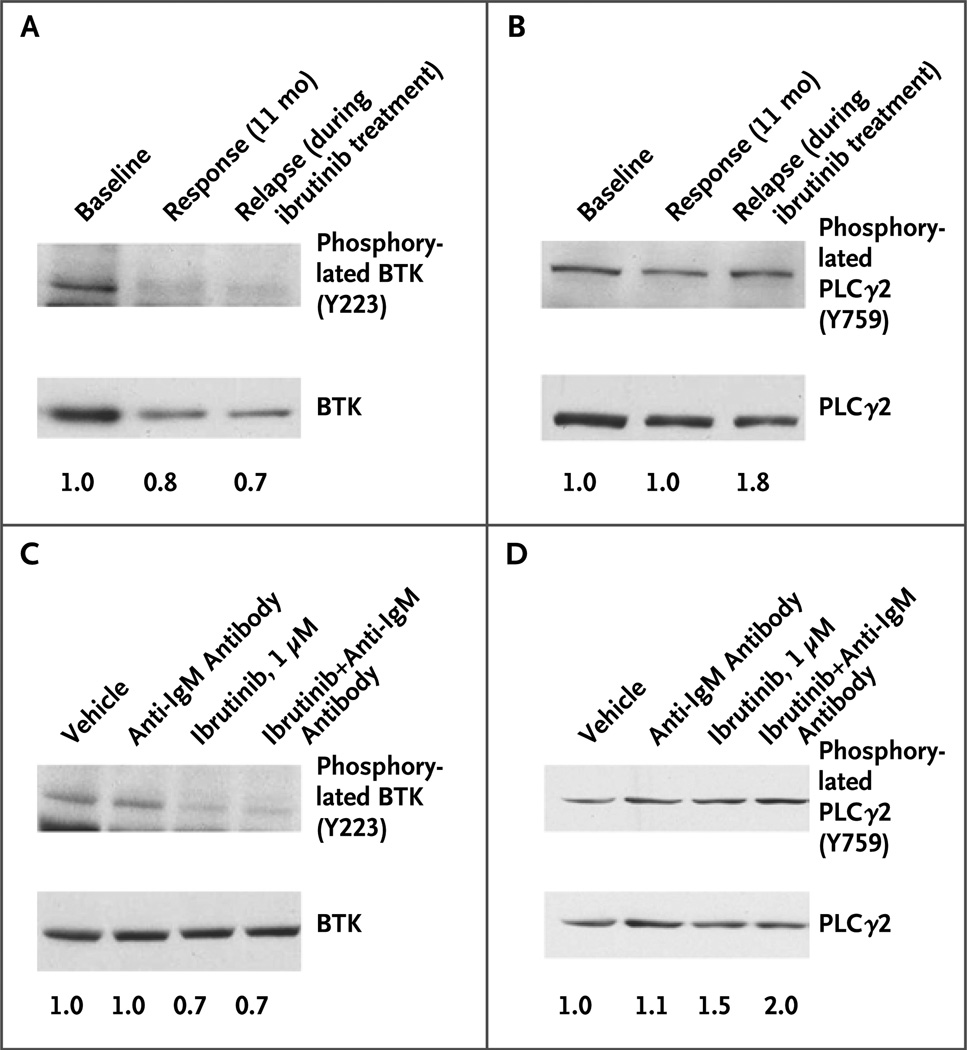
Results: We identified a cysteine-to-serine mutation in BTK at the binding site of ibrutinib in five patients and identified three distinct mutations in PLCγ2 in two patients. Functional analysis showed that the C481S mutation of BTK results in a protein that is only reversibly inhibited by ibrutinib. The R665W and L845F mutations in PLCγ2 are both potentially gain-of-function mutations that lead to autonomous B-cell-receptor activity. These mutations were not found in any of the patients with prolonged lymphocytosis who were taking ibrutinib.
Conclusions: Resistance to the irreversible BTK inhibitor ibrutinib often involves mutation of a cysteine residue where ibrutinib binding occurs. This finding, combined with two additional mutations in PLCγ2 that are immediately downstream of BTK, underscores the importance of the B-cell-receptor pathway in the mechanism of action of ibrutinib in CLL. (Funded by the National Cancer Institute and others.).
Figures
Comment in
-
Targeted therapies: ibrutinib resonates with us.Nat Rev Clin Oncol. 2014 Jul;11(7):380. doi: 10.1038/nrclinonc.2014.105. Epub 2014 Jun 17. Nat Rev Clin Oncol. 2014. PMID: 24935013 No abstract available.
References
-
- Dal Porto JM, Gauld SB, Merrell KT, Mills D, Pugh-Bernard AE, Cambier J. B cell antigen receptor signaling 101. Mol Immunol. 2004;41:599–613. - PubMed
-
- Deglesne PA, Chevallier N, Letestu R, et al. Survival response to B-cell receptor ligation is restricted to progressive chronic lymphocytic leukemia cells irrespective of Zap70 expression. Cancer Res. 2006;66:7158–7166. - PubMed
-
- Bernal A, Pastore RD, Asgary Z, et al. Survival of leukemic B cells promoted by engagement of the antigen receptor. Blood. 2001;98:3050–3057. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases