

A pan-cancer proteomic perspective on The Cancer Genome Atlas
- PMID: 24871328
- PMCID: PMC4109726
- DOI: 10.1038/ncomms4887
A pan-cancer proteomic perspective on The Cancer Genome Atlas
Erratum in
-
Corrigendum: A pan-cancer proteomic perspective on The Cancer Genome Atlas.Nat Commun. 2015 Jan 28;6:4852. doi: 10.1038/ncomms5852. Nat Commun. 2015. PMID: 25629879 No abstract available.
Abstract
Protein levels and function are poorly predicted by genomic and transcriptomic analysis of patient tumours. Therefore, direct study of the functional proteome has the potential to provide a wealth of information that complements and extends genomic, epigenomic and transcriptomic analysis in The Cancer Genome Atlas (TCGA) projects. Here we use reverse-phase protein arrays to analyse 3,467 patient samples from 11 TCGA 'Pan-Cancer' diseases, using 181 high-quality antibodies that target 128 total proteins and 53 post-translationally modified proteins. The resultant proteomic data are integrated with genomic and transcriptomic analyses of the same samples to identify commonalities, differences, emergent pathways and network biology within and across tumour lineages. In addition, tissue-specific signals are reduced computationally to enhance biomarker and target discovery spanning multiple tumour lineages. This integrative analysis, with an emphasis on pathways and potentially actionable proteins, provides a framework for determining the prognostic, predictive and therapeutic relevance of the functional proteome.
Conflict of interest statement
The authors have no conflicts of interest to declare.
Figures
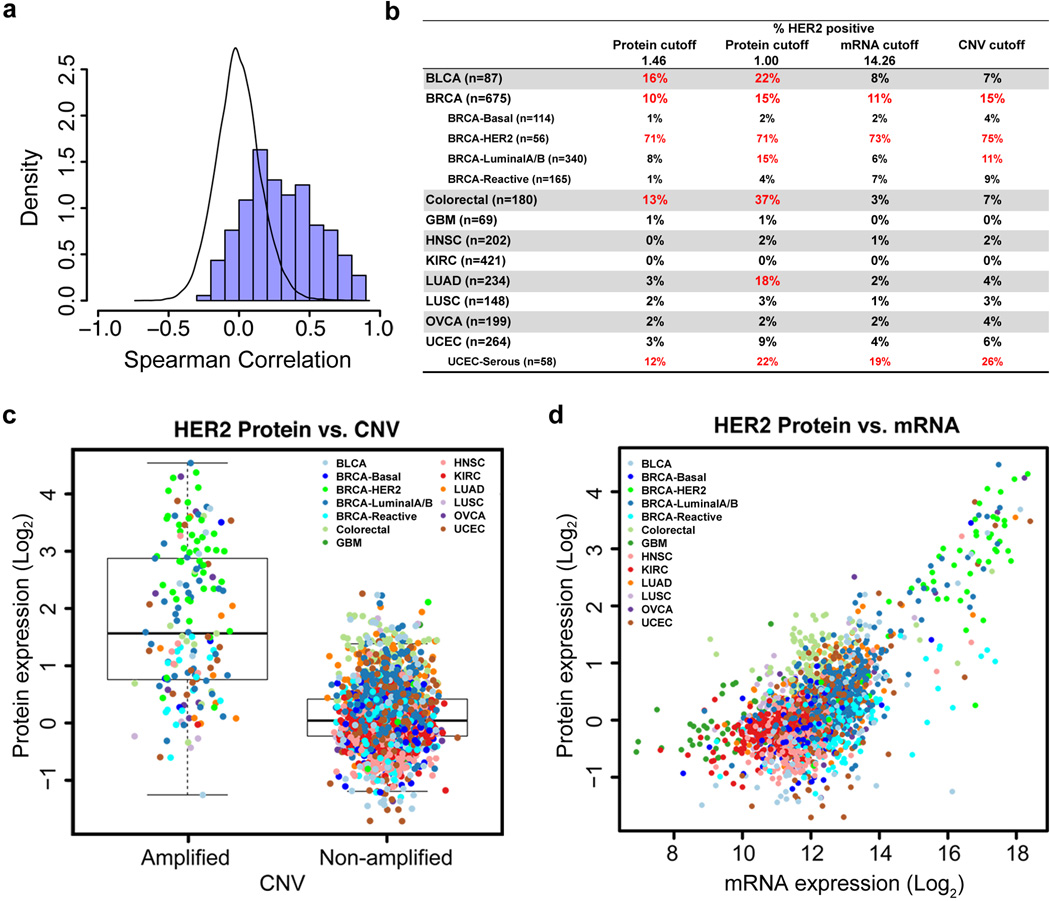
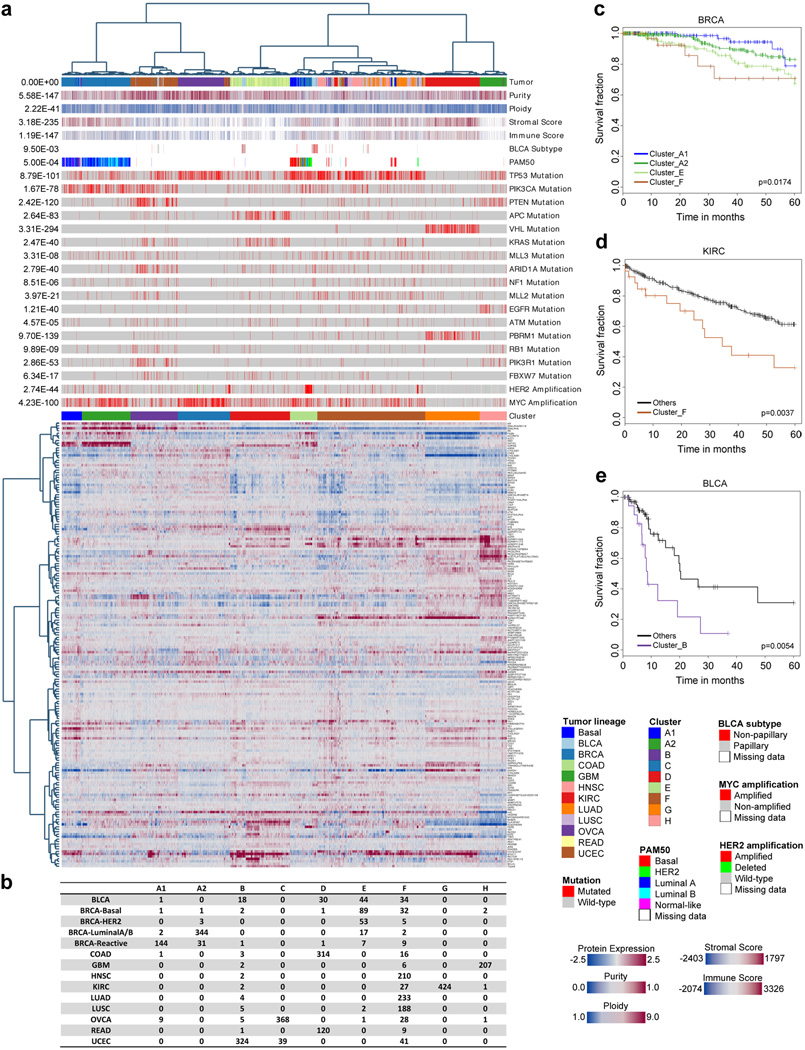
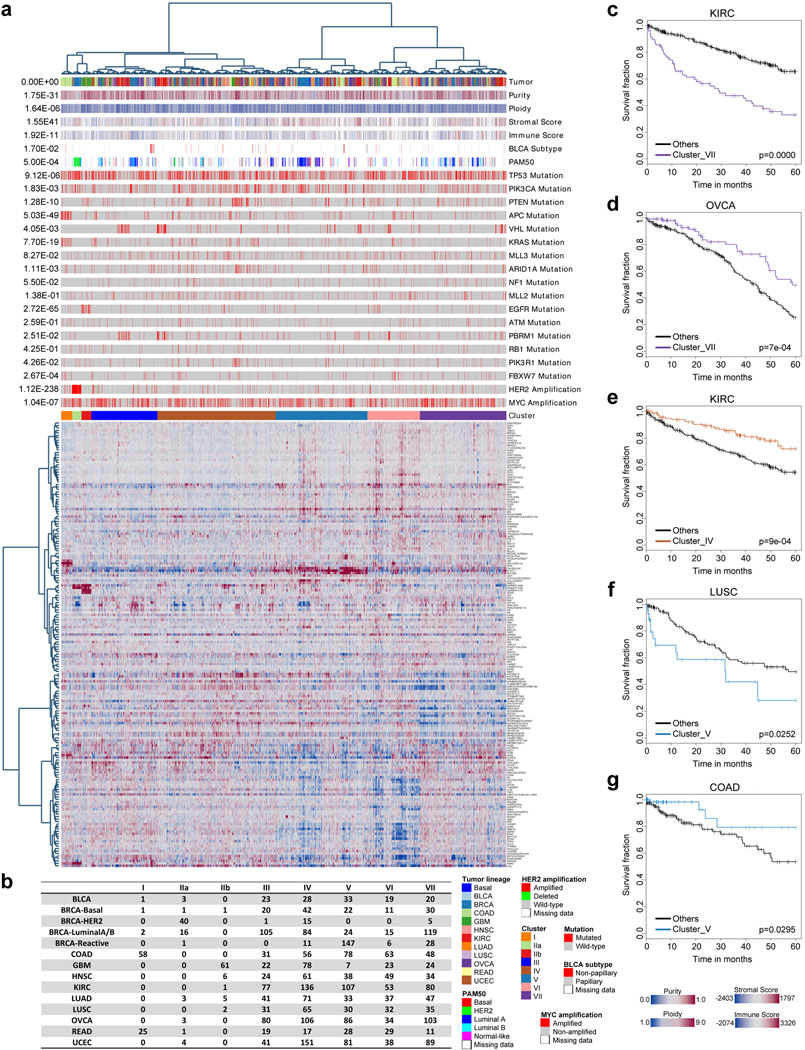
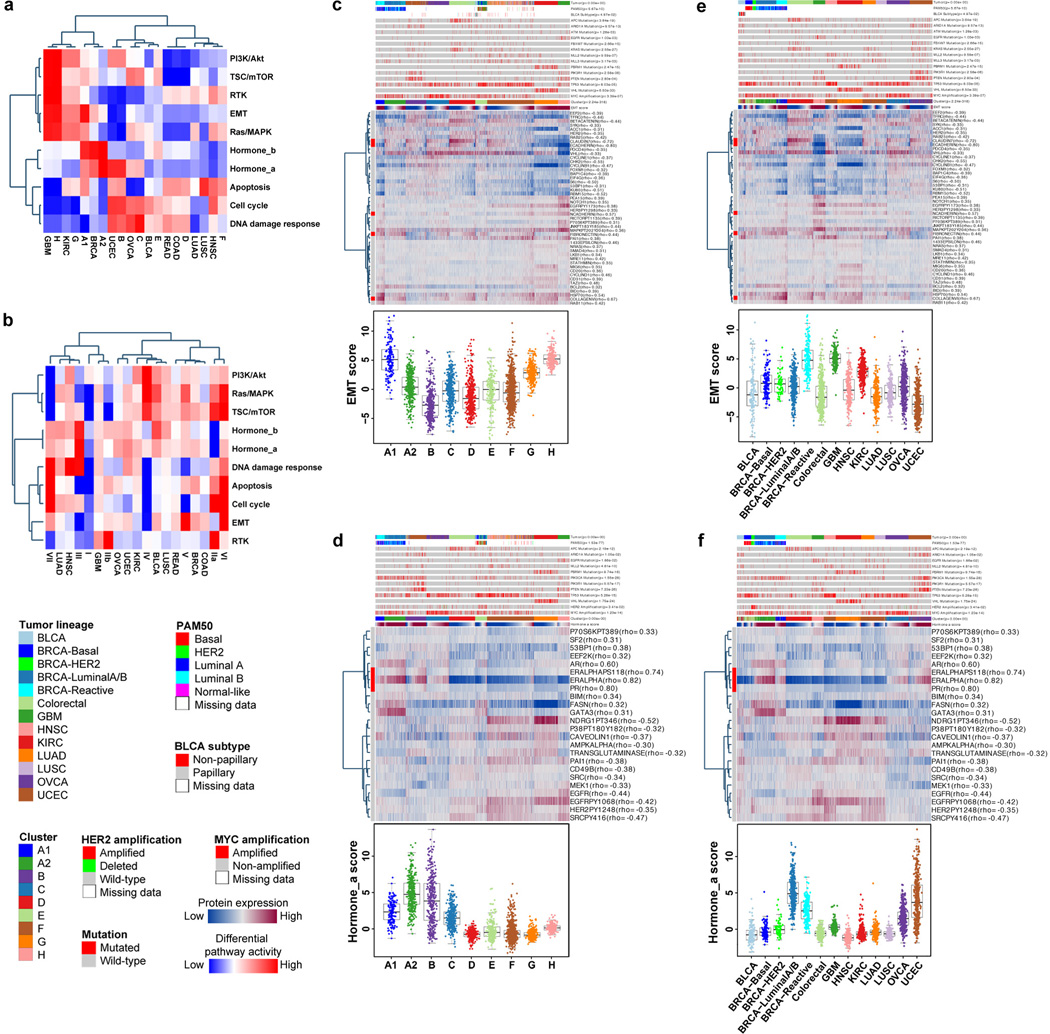
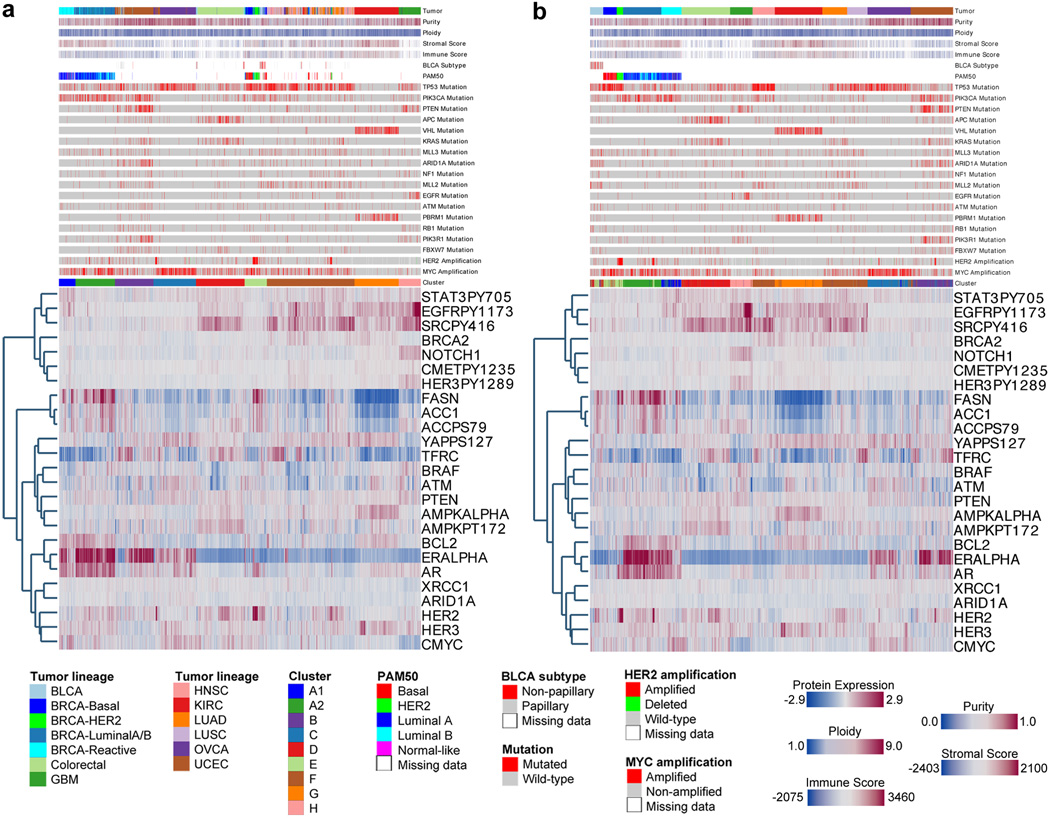
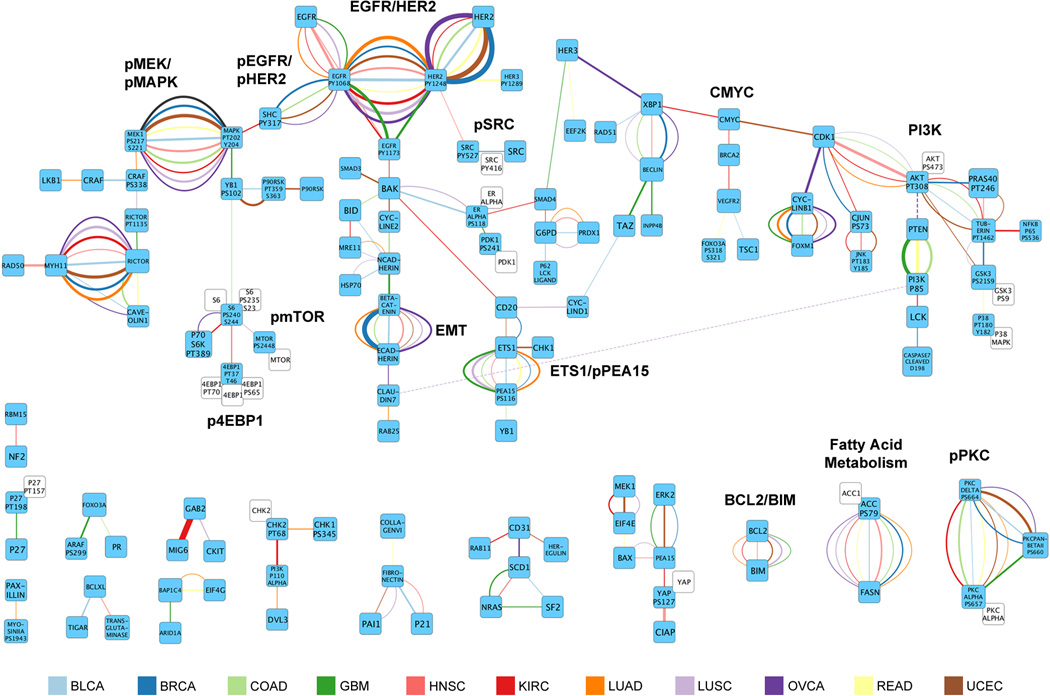
References
-
- Shankavaram UT, et al. Transcript and protein expression profiles of the NCI-60 cancer cell panel: an integromic microarray study. Molecular cancer therapeutics. 2007;6:820–832. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- P30 CA016672/CA/NCI NIH HHS/United States
- P50 CA100632/CA/NCI NIH HHS/United States
- MC_UP_1302/1/MRC_/Medical Research Council/United Kingdom
- U54 CA112970/CA/NCI NIH HHS/United States
- TCGA CA143883/CA/NCI NIH HHS/United States
- U24 CA143883/CA/NCI NIH HHS/United States
- NCI P50CA70907/CA/NCI NIH HHS/United States
- P50 CA098258/CA/NCI NIH HHS/United States
- U01 CA168394/CA/NCI NIH HHS/United States
- P50 CA083639/CA/NCI NIH HHS/United States
- NCI U54 CA112970/CA/NCI NIH HHS/United States
- P50 CA070907/CA/NCI NIH HHS/United States
- U01 CA176284/CA/NCI NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
