

Ribosomal protein mutations induce autophagy through S6 kinase inhibition of the insulin pathway
- PMID: 24875531
- PMCID: PMC4038485
- DOI: 10.1371/journal.pgen.1004371
Ribosomal protein mutations induce autophagy through S6 kinase inhibition of the insulin pathway
Abstract
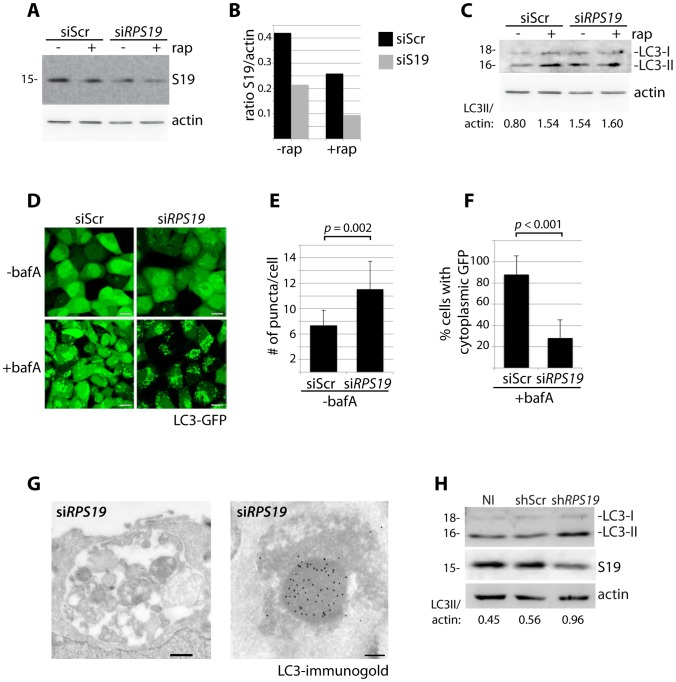
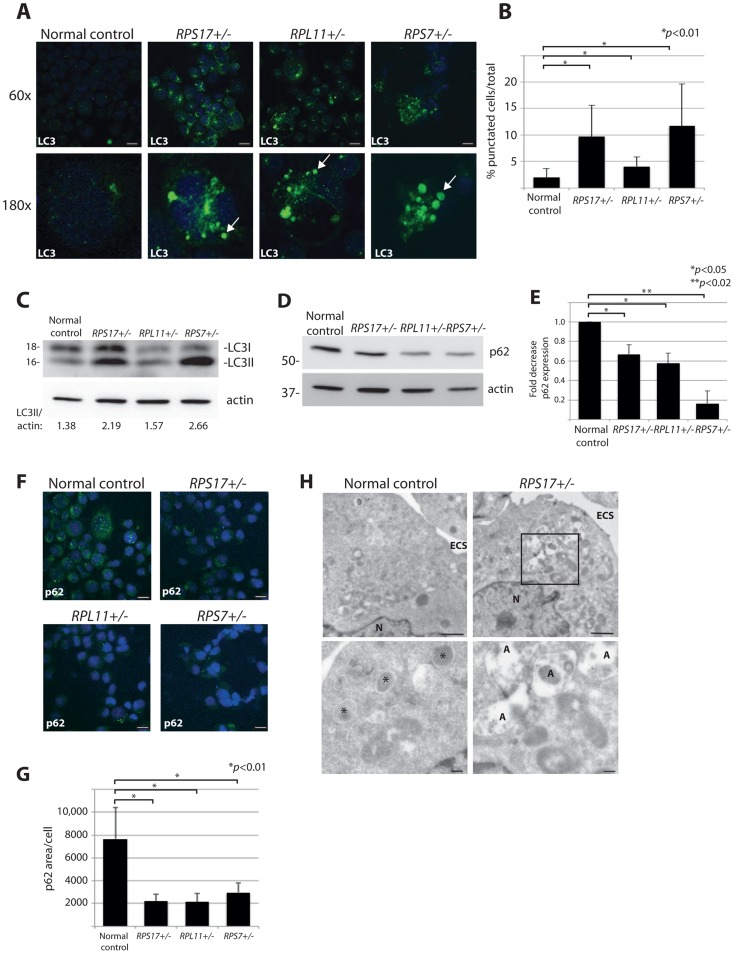
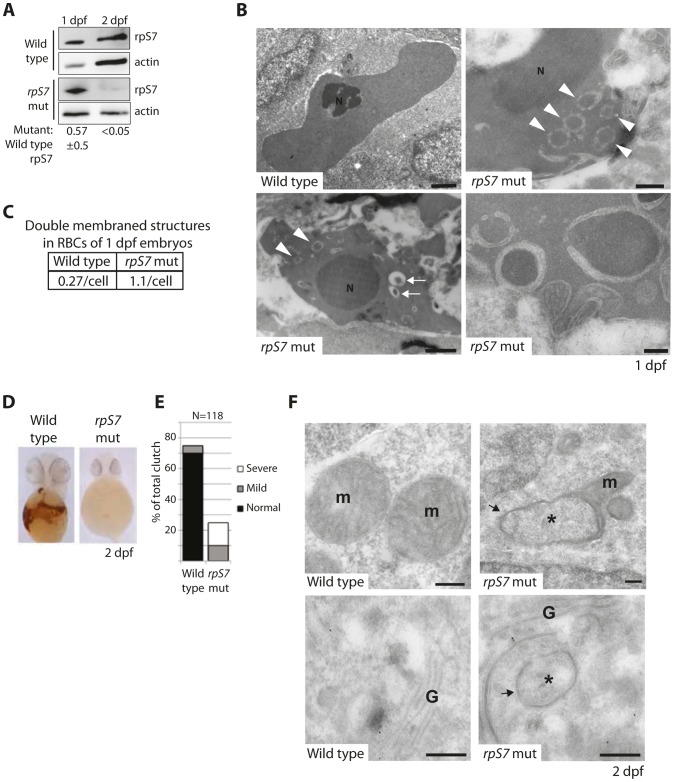
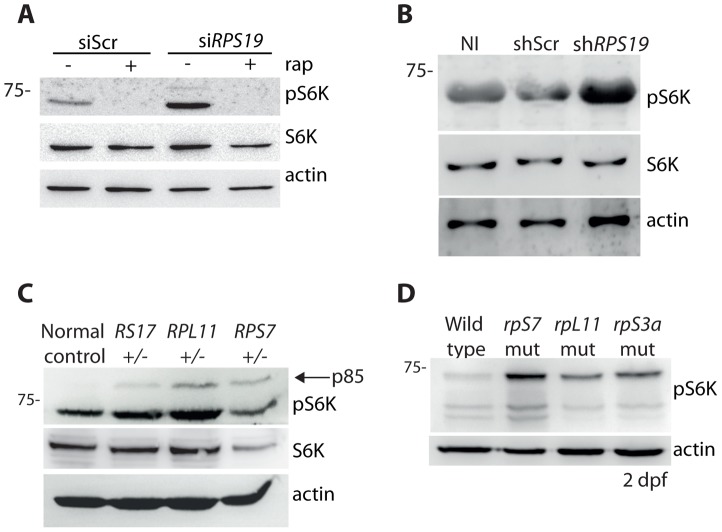
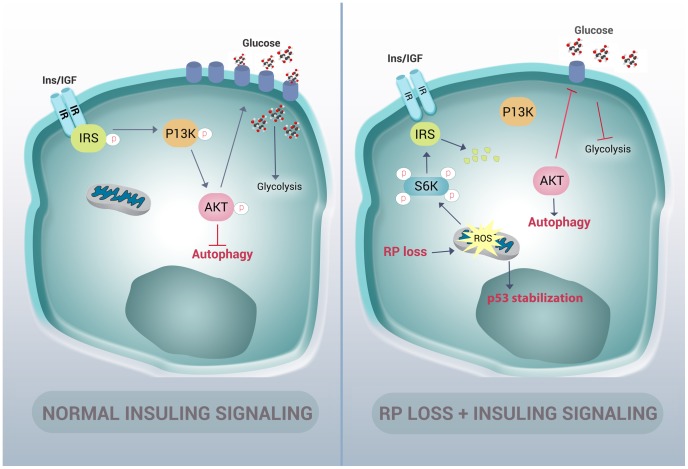
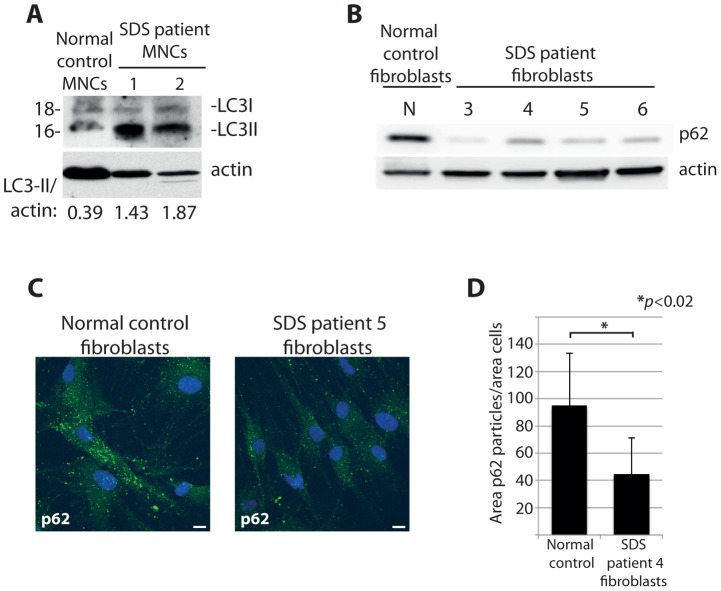
Mutations affecting the ribosome lead to several diseases known as ribosomopathies, with phenotypes that include growth defects, cytopenia, and bone marrow failure. Diamond-Blackfan anemia (DBA), for example, is a pure red cell aplasia linked to the mutation of ribosomal protein (RP) genes. Here we show the knock-down of the DBA-linked RPS19 gene induces the cellular self-digestion process of autophagy, a pathway critical for proper hematopoiesis. We also observe an increase of autophagy in cells derived from DBA patients, in CD34+ erythrocyte progenitor cells with RPS19 knock down, in the red blood cells of zebrafish embryos with RP-deficiency, and in cells from patients with Shwachman-Diamond syndrome (SDS). The loss of RPs in all these models results in a marked increase in S6 kinase phosphorylation that we find is triggered by an increase in reactive oxygen species (ROS). We show that this increase in S6 kinase phosphorylation inhibits the insulin pathway and AKT phosphorylation activity through a mechanism reminiscent of insulin resistance. While stimulating RP-deficient cells with insulin reduces autophagy, antioxidant treatment reduces S6 kinase phosphorylation, autophagy, and stabilization of the p53 tumor suppressor. Our data suggest that RP loss promotes the aberrant activation of both S6 kinase and p53 by increasing intracellular ROS levels. The deregulation of these signaling pathways is likely playing a major role in the pathophysiology of ribosomopathies.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures
Similar articles
-
Ribosomal Protein Mutations Result in Constitutive p53 Protein Degradation through Impairment of the AKT Pathway.PLoS Genet. 2015 Jul 1;11(7):e1005326. doi: 10.1371/journal.pgen.1005326. eCollection 2015 Jul. PLoS Genet. 2015. PMID: 26132763 Free PMC article.
-
Reduced EIF6 dosage attenuates TP53 activation in models of Shwachman-Diamond syndrome.J Clin Invest. 2025 Feb 18;135(8):e187778. doi: 10.1172/JCI187778. eCollection 2025 Apr 15. J Clin Invest. 2025. PMID: 39964763 Free PMC article.
-
Ribosomal protein deficiency causes Tp53-independent erythropoiesis failure in zebrafish.Int J Biochem Cell Biol. 2014 Apr;49:1-7. doi: 10.1016/j.biocel.2014.01.006. Epub 2014 Jan 11. Int J Biochem Cell Biol. 2014. PMID: 24417973
-
Non-Diamond Blackfan anemia disorders of ribosome function: Shwachman Diamond syndrome and 5q- syndrome.Semin Hematol. 2011 Apr;48(2):136-43. doi: 10.1053/j.seminhematol.2011.01.002. Semin Hematol. 2011. PMID: 21435510 Free PMC article. Review.
-
Do ribosomopathies explain some cases of common variable immunodeficiency?Clin Exp Immunol. 2011 Jan;163(1):96-103. doi: 10.1111/j.1365-2249.2010.04280.x. Epub 2010 Nov 9. Clin Exp Immunol. 2011. PMID: 21062271 Free PMC article. Review.
Cited by
-
Why is an energy metabolic defect the common outcome in BMFS?Cell Cycle. 2016 Oct;15(19):2571-2575. doi: 10.1080/15384101.2016.1218103. Epub 2016 Aug 11. Cell Cycle. 2016. PMID: 27579499 Free PMC article.
-
A memory of eS25 loss drives resistance phenotypes.Nucleic Acids Res. 2020 Jul 27;48(13):7279-7297. doi: 10.1093/nar/gkaa444. Nucleic Acids Res. 2020. PMID: 32463448 Free PMC article.
-
The Molecular and Genetic Mechanisms of Inherited Bone Marrow Failure Syndromes: The Role of Inflammatory Cytokines in Their Pathogenesis.Biomolecules. 2023 Aug 16;13(8):1249. doi: 10.3390/biom13081249. Biomolecules. 2023. PMID: 37627314 Free PMC article. Review.
-
Ribosomopathies: how a common root can cause a tree of pathologies.Dis Model Mech. 2015 Sep;8(9):1013-26. doi: 10.1242/dmm.020529. Dis Model Mech. 2015. PMID: 26398160 Free PMC article. Review.
-
Role of ribosomal protein mutations in tumor development (Review).Int J Oncol. 2016 Apr;48(4):1313-24. doi: 10.3892/ijo.2016.3387. Epub 2016 Feb 9. Int J Oncol. 2016. PMID: 26892688 Free PMC article. Review.
References
-
- Lipton JM, Kudisch M, Gross R, Nathan DG (1986) Defective erythroid progenitor differentiation system in congenital hypoplastic (Diamond-Blackfan) anemia. Blood 67: 962–968. - PubMed
-
- Ohene-Abuakwa Y, Orfali KA, Marius C, Ball SE (2005) Two-phase culture in Diamond Blackfan anemia: localization of erythroid defect. Blood 105: 838–846. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
