

A community effort to assess and improve drug sensitivity prediction algorithms
- PMID: 24880487
- PMCID: PMC4547623
- DOI: 10.1038/nbt.2877
A community effort to assess and improve drug sensitivity prediction algorithms
Abstract
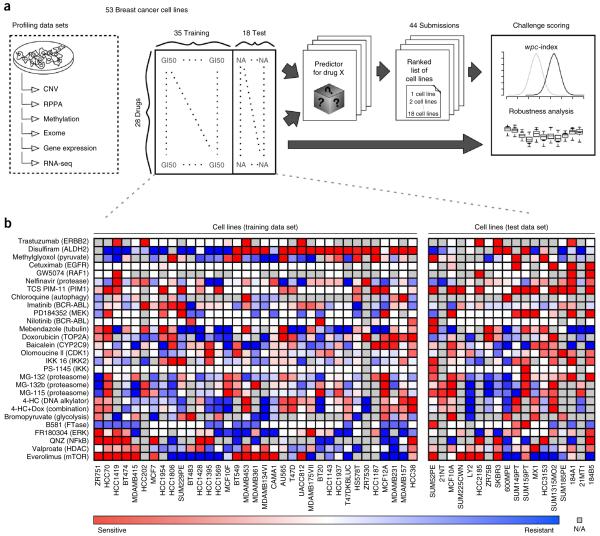
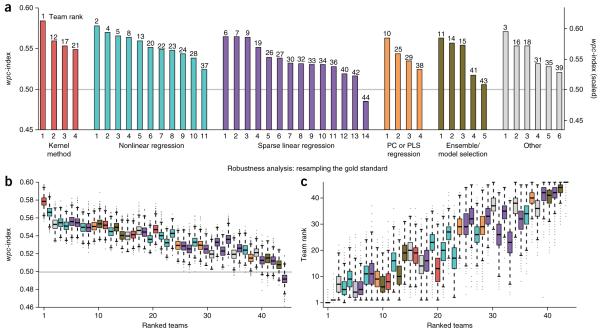
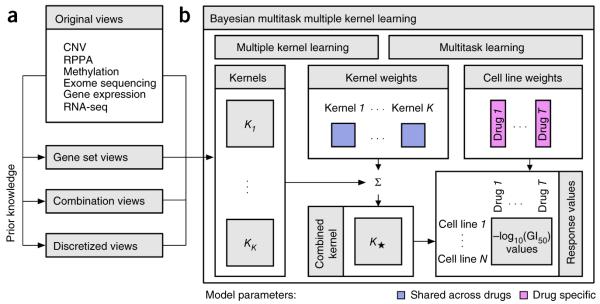
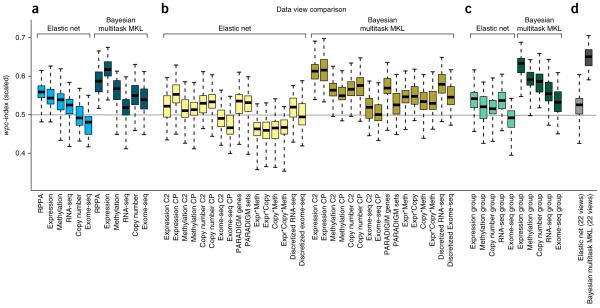
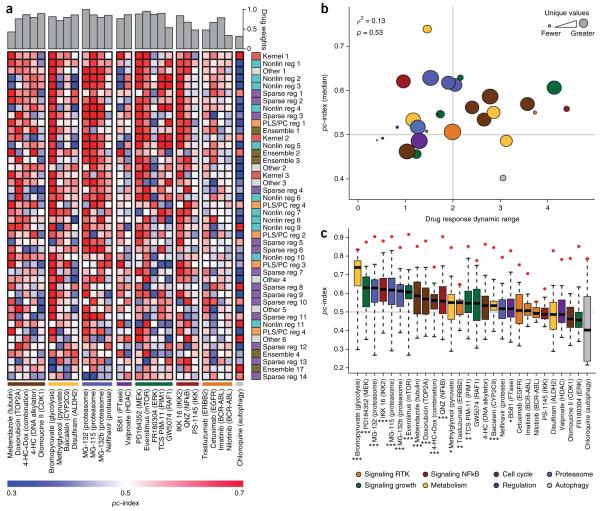
Predicting the best treatment strategy from genomic information is a core goal of precision medicine. Here we focus on predicting drug response based on a cohort of genomic, epigenomic and proteomic profiling data sets measured in human breast cancer cell lines. Through a collaborative effort between the National Cancer Institute (NCI) and the Dialogue on Reverse Engineering Assessment and Methods (DREAM) project, we analyzed a total of 44 drug sensitivity prediction algorithms. The top-performing approaches modeled nonlinear relationships and incorporated biological pathway information. We found that gene expression microarrays consistently provided the best predictive power of the individual profiling data sets; however, performance was increased by including multiple, independent data sets. We discuss the innovations underlying the top-performing methodology, Bayesian multitask MKL, and we provide detailed descriptions of all methods. This study establishes benchmarks for drug sensitivity prediction and identifies approaches that can be leveraged for the development of new methods.
Figures
References
Publication types
MeSH terms
Substances
Associated data
- Actions
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
