

Computationally driven deletion of broadly distributed T cell epitopes in a biotherapeutic candidate
- PMID: 24880662
- PMCID: PMC4234684
- DOI: 10.1007/s00018-014-1652-x
Computationally driven deletion of broadly distributed T cell epitopes in a biotherapeutic candidate
Abstract
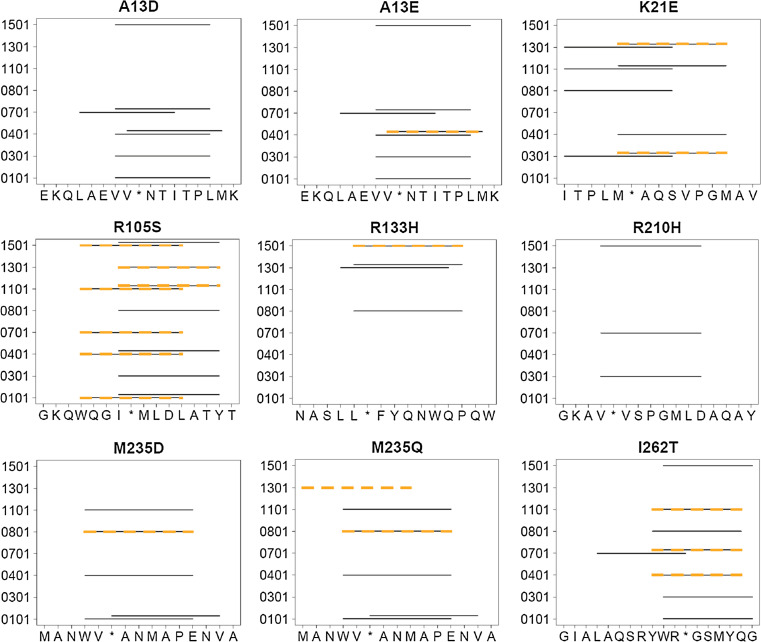
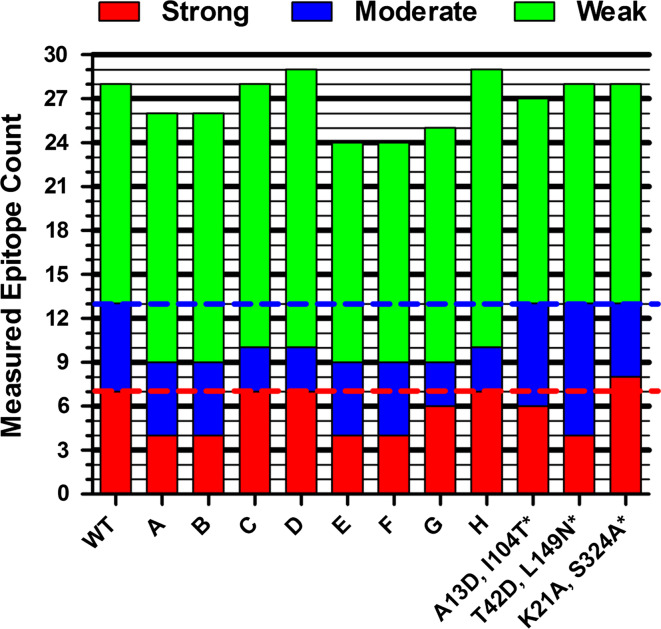
Biotherapeutics are subject to immune surveillance within the body, and anti-biotherapeutic immune responses can compromise drug efficacy and patient safety. Initial development of targeted antidrug immune memory is coordinated by T cell recognition of immunogenic subsequences, termed "T cell epitopes." Biotherapeutics may therefore be deimmunized by mutating key residues within cognate epitopes, but there exist complex trade-offs between immunogenicity, mutational load, and protein structure-function. Here, a protein deimmunization algorithm has been applied to P99 beta-lactamase, a component of antibody-directed enzyme prodrug therapies. The algorithm, integer programming for immunogenic proteins, seamlessly integrates computational prediction of T cell epitopes with both 1- and 2-body sequence potentials that assess protein tolerance to epitope-deleting mutations. Compared to previously deimmunized P99 variants, which bore only one or two mutations, the enzymes designed here contain 4-5 widely distributed substitutions. As a result, they exhibit broad reductions in major histocompatibility complex recognition. Despite their high mutational loads and markedly reduced immunoreactivity, all eight engineered variants possessed wild-type or better catalytic activity. Thus, the protein design algorithm is able to disrupt broadly distributed epitopes while maintaining protein function. As a result, this computational tool may prove useful in expanding the repertoire of next-generation biotherapeutics.
Conflict of interest statement
Karl E. Griswold and Chris Bailey-Kellogg are Dartmouth faculty and co-members of Stealth Biologics, LLC, a Delaware biotechnology company. They acknowledge that there is a potential conflict of interest related to their association with this company, and they hereby affirm that the data presented in this paper is free of any bias. This work has been reviewed and approved as specified in these faculty members’ Dartmouth conflict of interest management plans. The remaining authors declare no conflict of interest.
Figures



Similar articles
-
Design and analysis of immune-evading enzymes for ADEPT therapy.Protein Eng Des Sel. 2012 Oct;25(10):613-23. doi: 10.1093/protein/gzs044. Epub 2012 Aug 16. Protein Eng Des Sel. 2012. PMID: 22898588 Free PMC article.
-
Protein deimmunization via structure-based design enables efficient epitope deletion at high mutational loads.Biotechnol Bioeng. 2015 Jul;112(7):1306-18. doi: 10.1002/bit.25554. Epub 2015 Feb 23. Biotechnol Bioeng. 2015. PMID: 25655032 Free PMC article.
-
EpiSweep: Computationally Driven Reengineering of Therapeutic Proteins to Reduce Immunogenicity While Maintaining Function.Methods Mol Biol. 2017;1529:375-398. doi: 10.1007/978-1-4939-6637-0_20. Methods Mol Biol. 2017. PMID: 27914063 Free PMC article.
-
Design and engineering of deimmunized biotherapeutics.Curr Opin Struct Biol. 2016 Aug;39:79-88. doi: 10.1016/j.sbi.2016.06.003. Epub 2016 Jun 17. Curr Opin Struct Biol. 2016. PMID: 27322891 Free PMC article. Review.
-
Better Epitope Discovery, Precision Immune Engineering, and Accelerated Vaccine Design Using Immunoinformatics Tools.Front Immunol. 2020 Apr 7;11:442. doi: 10.3389/fimmu.2020.00442. eCollection 2020. Front Immunol. 2020. PMID: 32318055 Free PMC article. Review.
Cited by
-
Discovery-stage identification of drug-like antibodies using emerging experimental and computational methods.MAbs. 2021 Jan-Dec;13(1):1895540. doi: 10.1080/19420862.2021.1895540. MAbs. 2021. PMID: 34313532 Free PMC article. Review.
-
Poor correlation between T-cell activation assays and HLA-DR binding prediction algorithms in an immunogenic fragment of Pseudomonas exotoxin A.J Immunol Methods. 2015 Oct;425:10-20. doi: 10.1016/j.jim.2015.06.003. Epub 2015 Jun 6. J Immunol Methods. 2015. PMID: 26056938 Free PMC article.
-
Role of HLA-DP in the Presentation of Epitopes from the Truncated Bacterial PE38 Immunotoxin.AAPS J. 2017 Jan;19(1):117-129. doi: 10.1208/s12248-016-9986-y. Epub 2016 Oct 27. AAPS J. 2017. PMID: 27796910 Free PMC article.
-
Machine-guided dual-objective protein engineering for deimmunization and therapeutic functions.Cell Syst. 2025 Jul 16;16(7):101299. doi: 10.1016/j.cels.2025.101299. Epub 2025 Jun 3. Cell Syst. 2025. PMID: 40466641
-
QM/MM Description of Newly Selected Catalytic Bioscavengers Against Organophosphorus Compounds Revealed Reactivation Stimulus Mediated by Histidine Residue in the Acyl-Binding Loop.Front Pharmacol. 2018 Aug 3;9:834. doi: 10.3389/fphar.2018.00834. eCollection 2018. Front Pharmacol. 2018. PMID: 30123127 Free PMC article.
References
-
- Schellekens H. Immunogenicity of protein therapeutics, or how to make antibodies without T-cells. Inflamm Res. 2007;56:S351–S352.
-
- Schellekens H (2005) Factors influencing the immunogenicity of therapeutic proteins. Nephrol Dial Transplant 20(Suppl 6):vi3–vi9. doi:10.1093/ndt/gfh1092 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
