

Crystal cryocooling distorts conformational heterogeneity in a model Michaelis complex of DHFR
- PMID: 24882744
- PMCID: PMC4082491
- DOI: 10.1016/j.str.2014.04.016
Crystal cryocooling distorts conformational heterogeneity in a model Michaelis complex of DHFR
Abstract
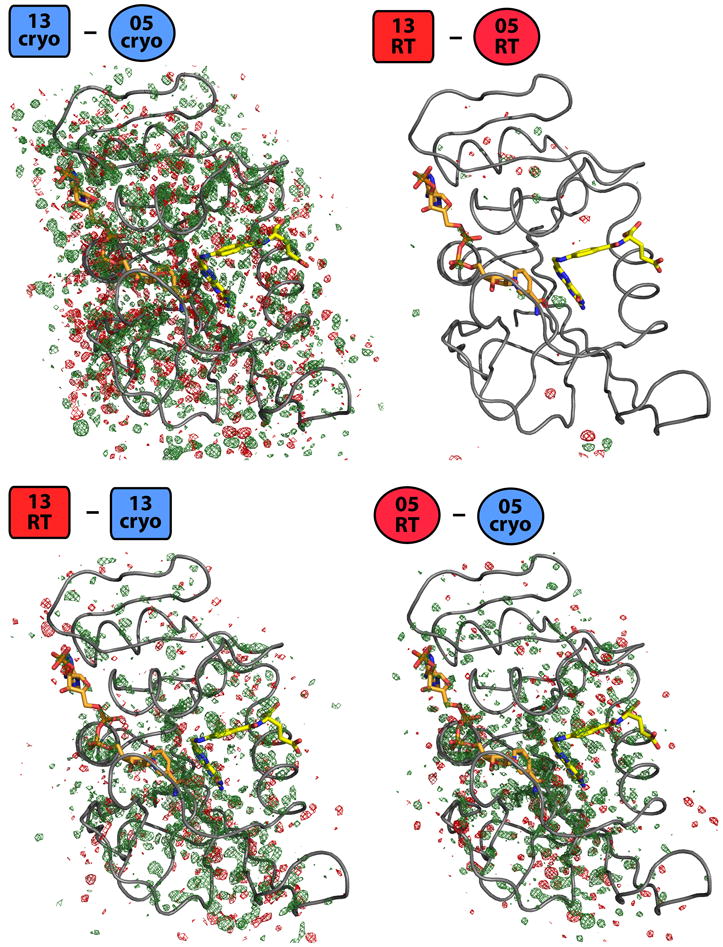
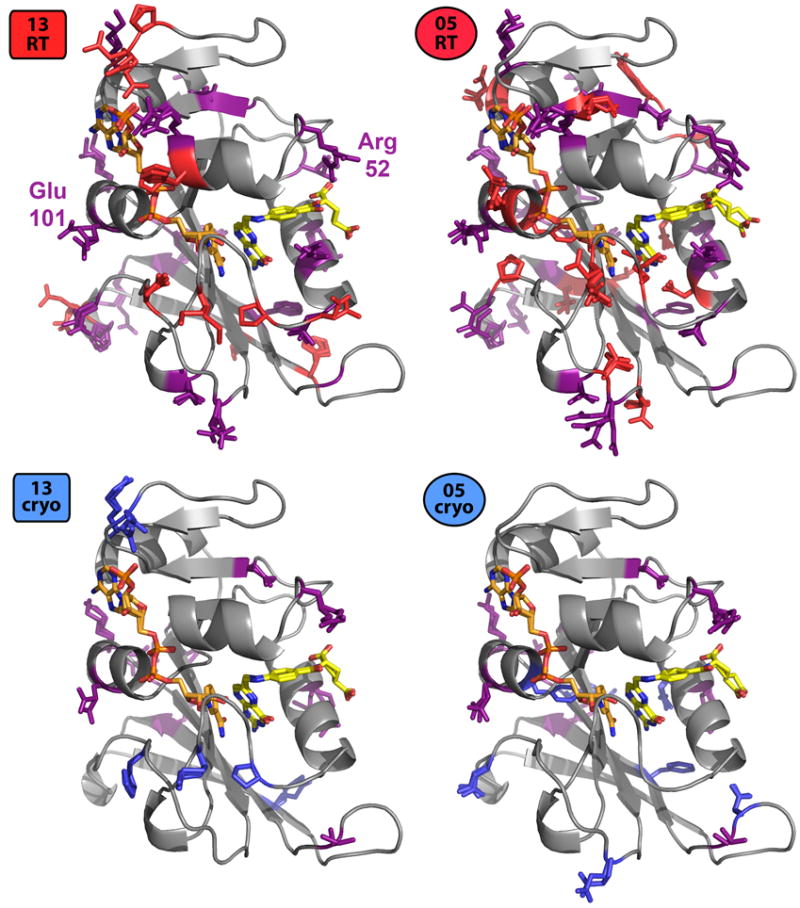
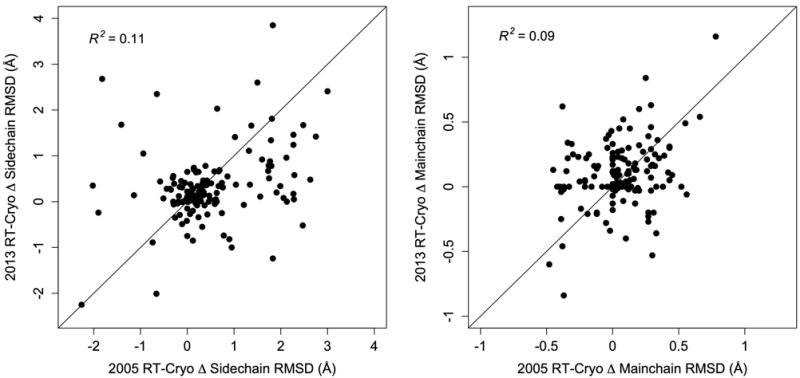
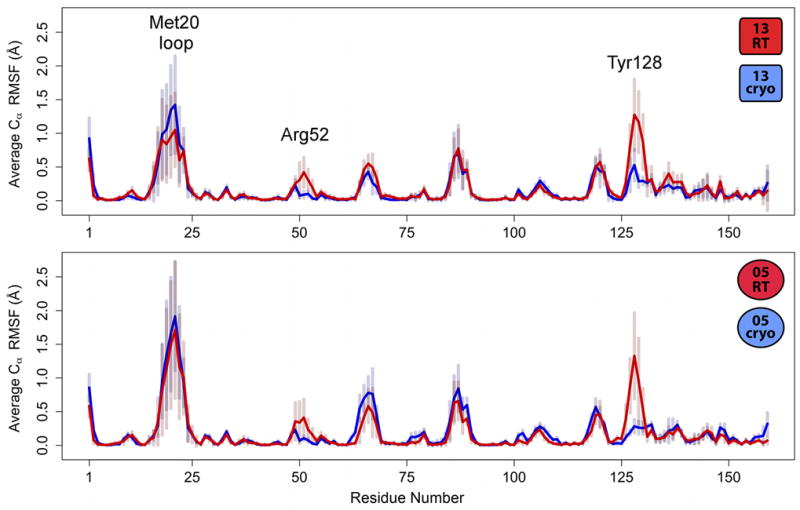
Most macromolecular X-ray structures are determined from cryocooled crystals, but it is unclear whether cryocooling distorts functionally relevant flexibility. Here we compare independently acquired pairs of high-resolution data sets of a model Michaelis complex of dihydrofolate reductase (DHFR), collected by separate groups at both room and cryogenic temperatures. These data sets allow us to isolate the differences between experimental procedures and between temperatures. Our analyses of multiconformer models and time-averaged ensembles suggest that cryocooling suppresses and otherwise modifies side-chain and main-chain conformational heterogeneity, quenching dynamic contact networks. Despite some idiosyncratic differences, most changes from room temperature to cryogenic temperature are conserved and likely reflect temperature-dependent solvent remodeling. Both cryogenic data sets point to additional conformations not evident in the corresponding room temperature data sets, suggesting that cryocooling does not merely trap preexisting conformational heterogeneity. Our results demonstrate that crystal cryocooling consistently distorts the energy landscape of DHFR, a paragon for understanding functional protein dynamics.
Copyright © 2014 Elsevier Ltd. All rights reserved.
Figures
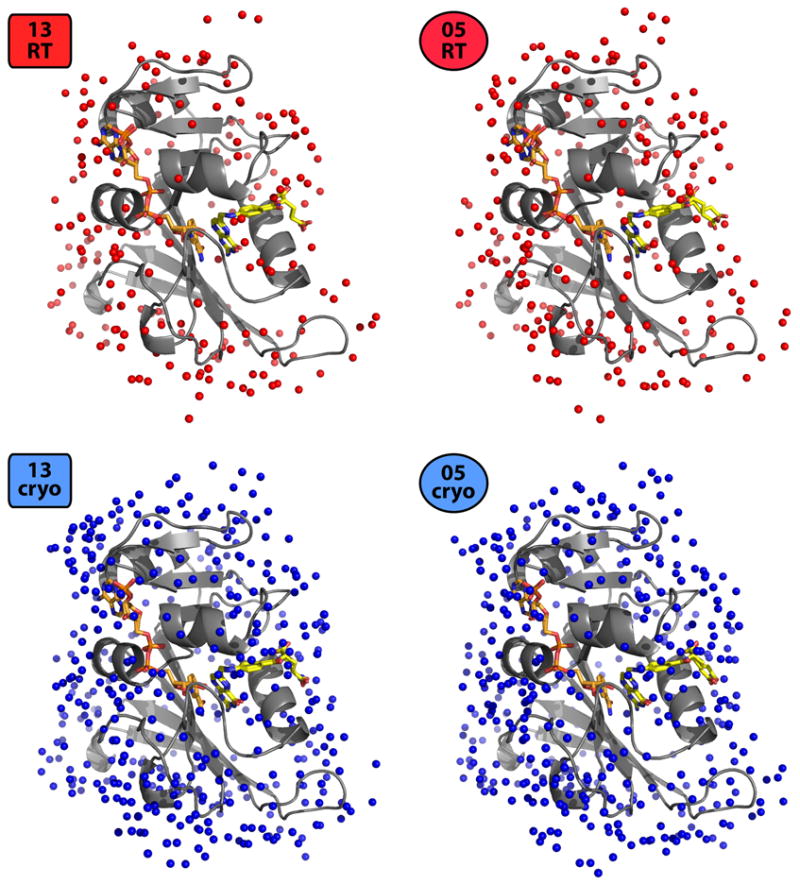
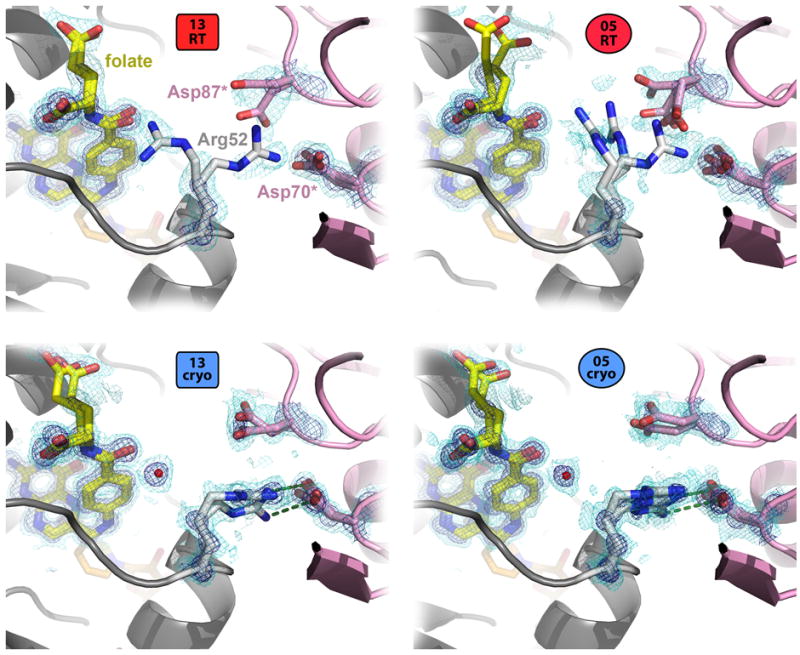
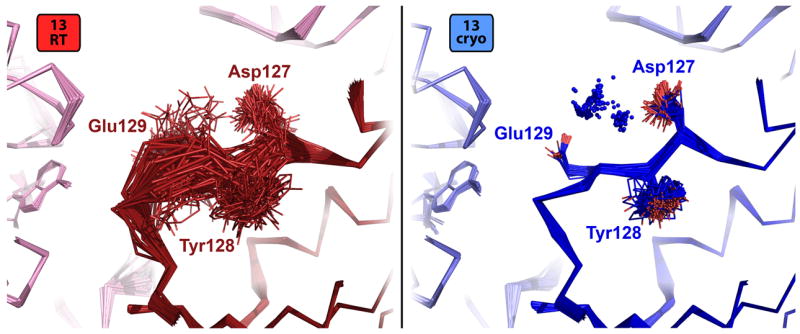
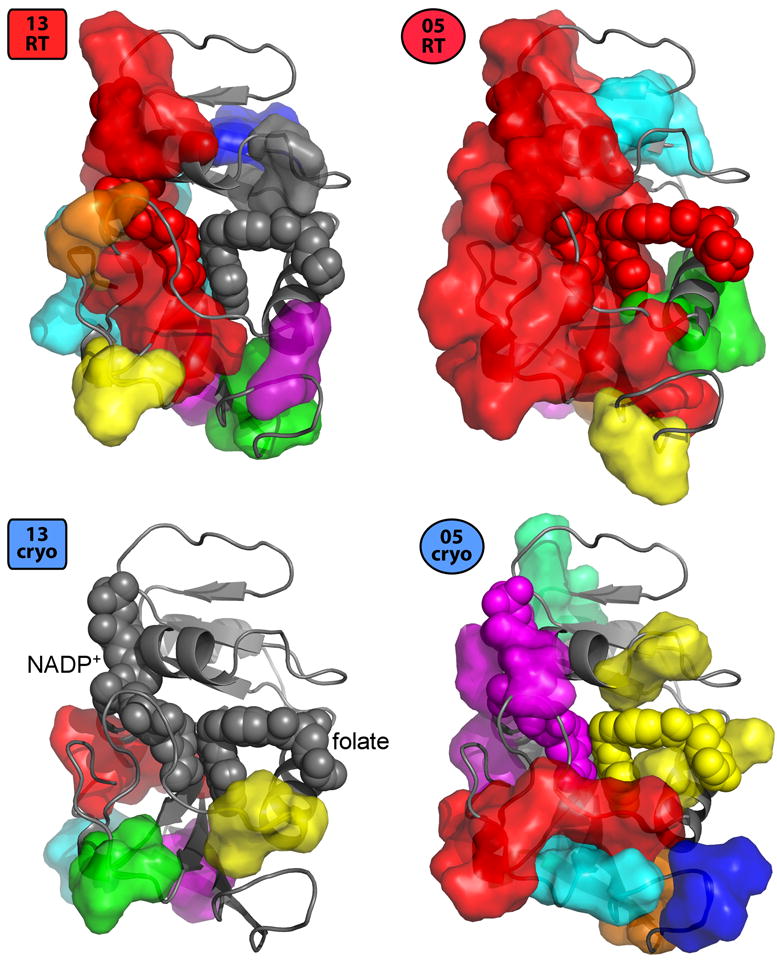
References
-
- Antal MA, Bode C, Csermely P. Perturbation waves in proteins and protein networks: applications of percolation and game theories in signaling and drug design. Current protein & peptide science. 2009;10:161–172. - PubMed
-
- Boehr DD, McElheny D, Dyson HJ, Wright PE. The dynamic energy landscape of dihydrofolate reductase catalysis. Science. 2006;313:1638–1642. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- R21 GM110580/GM/NIGMS NIH HHS/United States
- GM081879/GM/NIGMS NIH HHS/United States
- P50 GM081879/GM/NIGMS NIH HHS/United States
- R37 GM032415/GM/NIGMS NIH HHS/United States
- U54 GM094586/GM/NIGMS NIH HHS/United States
- U54GM094586/GM/NIGMS NIH HHS/United States
- GM092999/GM/NIGMS NIH HHS/United States
- P41GM103393/GM/NIGMS NIH HHS/United States
- OD009180/OD/NIH HHS/United States
- DP5 OD009180/OD/NIH HHS/United States
- GM110580/GM/NIGMS NIH HHS/United States
- R01 GM032415/GM/NIGMS NIH HHS/United States
- P41 GM103393/GM/NIGMS NIH HHS/United States
- GM32415/GM/NIGMS NIH HHS/United States
- R01 GM092999/GM/NIGMS NIH HHS/United States
