

First report of OPA1 screening in Greek patients with autosomal dominant optic atrophy and identification of a previously undescribed OPA1 mutation
- PMID: 24883014
- PMCID: PMC4037535
First report of OPA1 screening in Greek patients with autosomal dominant optic atrophy and identification of a previously undescribed OPA1 mutation
Abstract
Purpose: To describe the genotype-phenotype correlation in four Greek pedigrees with autosomal dominant optic atrophy (ADOA) and OPA1 mutations.
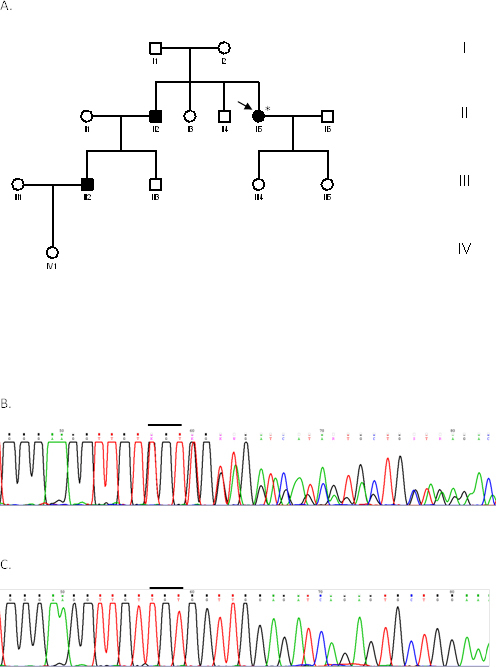
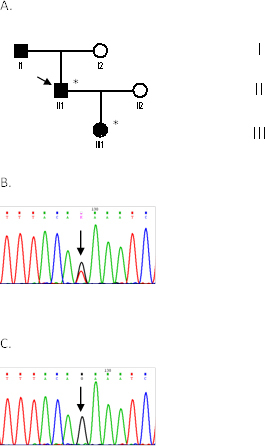
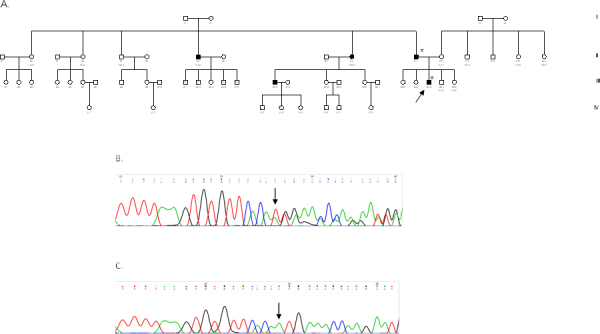
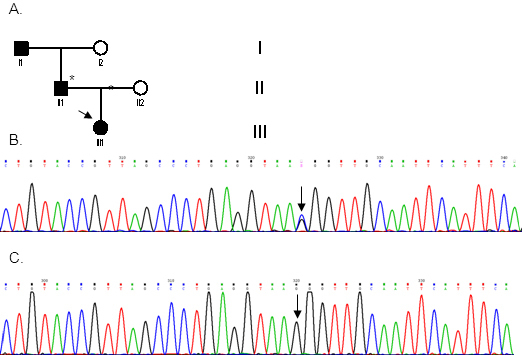
Methods: Seven patients from four unrelated families (F1, F2, F3, F4) were clinically assessed for visual acuity, color vision, ptosis, afferent pupillary defects, and visual fields and underwent orthoptic assessment, slit-lamp biomicroscopy, and fundus examination to establish their clinical status. Genomic DNA was extracted from peripheral blood samples from all participants. The coding region (exons 1-28), including the intron-exon boundaries of the OPA1 gene, was screened in the probands of the four families, as well as in seven additional family members (four affected and three unaffected) with PCR and direct DNA sequencing.
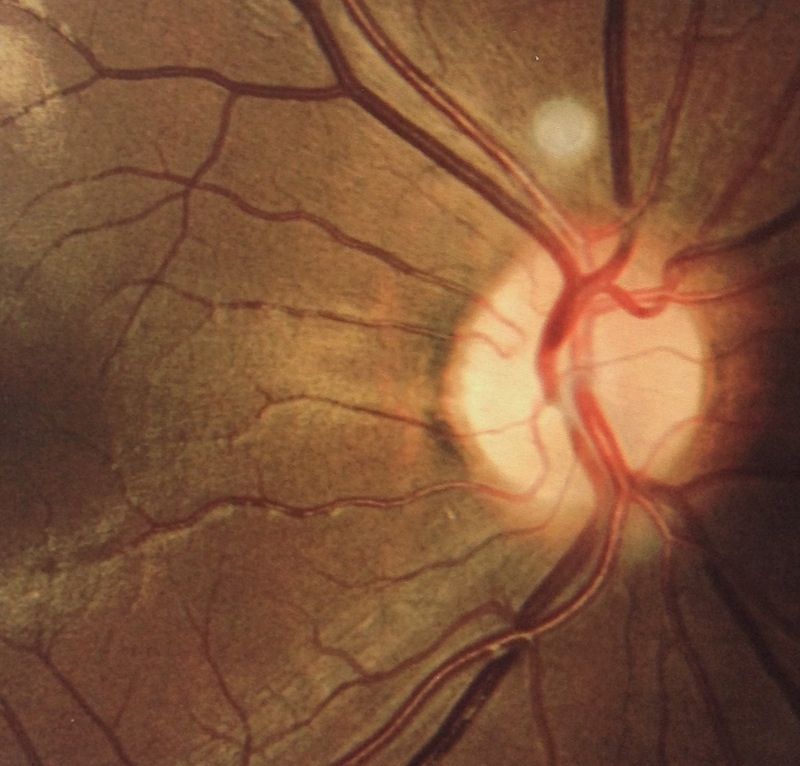
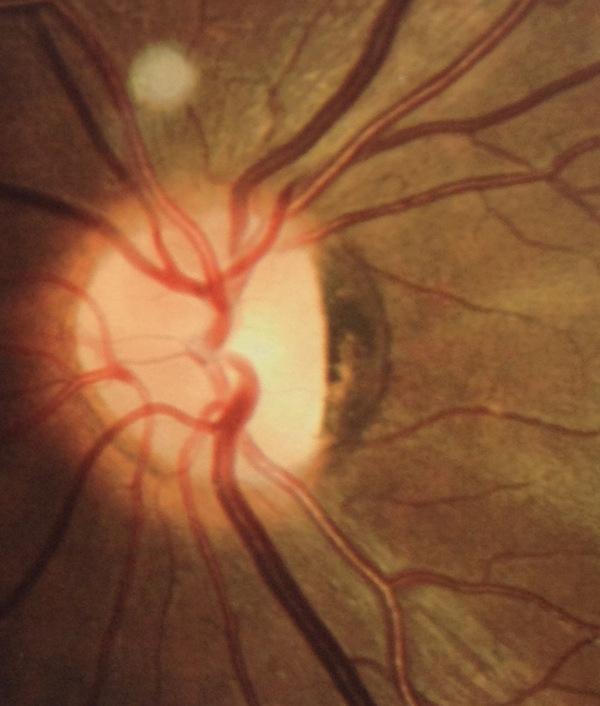
Results: All patients presented bilateral decrease in best-corrected visual acuity and temporal pallor of the optic disc. The visual fields of the adult patients showed characteristic scotomata. Other signs were present in some patients such as decreased color discrimination and a gray crescent within the neuroretinal rim. After the OPA1 gene was sequenced, a previously undescribed heterozygous splice-site mutation c.784-1G>T in intron 7 was detected in family F2. In families F1, F3, and F4, a previously reported in-frame deletion c.876_878delTGT/p.(Val294del), the frameshift c.2366delA/p.(Asn789Metfs*11), and splice-site c.1140+5G>C mutations were detected, respectively.
Conclusions: This is the first report of molecular characterization of Greek patients with ADOA. Our findings provide additional information regarding the genotype-phenotype correlation and establish the role of the OPA1 gene in Greek patients with ADOA.
Figures

Similar articles
-
Identification of two novel OPA1 mutations in Chinese families with autosomal dominant optic atrophy.Mol Vis. 2008;14:2451-7. Epub 2008 Dec 29. Mol Vis. 2008. PMID: 19112530 Free PMC article.
-
A comprehensive survey of mutations in the OPA1 gene in patients with autosomal dominant optic atrophy.Invest Ophthalmol Vis Sci. 2002 Jun;43(6):1715-24. Invest Ophthalmol Vis Sci. 2002. PMID: 12036970
-
Novel mutations in the OPA1 gene and associated clinical features in Japanese patients with optic atrophy.Ophthalmology. 2006 Mar;113(3):483-488.e1. doi: 10.1016/j.ophtha.2005.10.054. Ophthalmology. 2006. PMID: 16513463
-
Meta-analysis of genotype-phenotype analysis of OPA1 mutations in autosomal dominant optic atrophy.Mitochondrion. 2019 May;46:262-269. doi: 10.1016/j.mito.2018.07.006. Epub 2018 Aug 27. Mitochondrion. 2019. PMID: 30165240
-
Recessive optic atrophy, sensorimotor neuropathy and cataract associated with novel compound heterozygous mutations in OPA1.Mol Med Rep. 2016 Jul;14(1):33-40. doi: 10.3892/mmr.2016.5209. Epub 2016 May 4. Mol Med Rep. 2016. PMID: 27150940 Free PMC article. Review.
Cited by
-
Wolfram Syndrome: A case report of two sisters Wolfram Syndrome: Case report of two sisters.Am J Ophthalmol Case Rep. 2022 Mar 1;26:101452. doi: 10.1016/j.ajoc.2022.101452. eCollection 2022 Jun. Am J Ophthalmol Case Rep. 2022. PMID: 35252627 Free PMC article.
-
The OPA1 Gene Mutations Are Frequent in Han Chinese Patients with Suspected Optic Neuropathy.Mol Neurobiol. 2017 Apr;54(3):1622-1630. doi: 10.1007/s12035-016-9771-z. Epub 2016 Feb 11. Mol Neurobiol. 2017. PMID: 26867657
-
OPA1 Dominant Optic Atrophy: Diagnostic Approach in the Pediatric Population.Curr Issues Mol Biol. 2023 Jan 5;45(1):465-478. doi: 10.3390/cimb45010030. Curr Issues Mol Biol. 2023. PMID: 36661516 Free PMC article.
-
Mutation Spectrum of the ABCA4 Gene in a Greek Cohort with Stargardt Disease: Identification of Novel Mutations and Evidence of Three Prevalent Mutated Alleles.J Ophthalmol. 2018 Apr 30;2018:5706142. doi: 10.1155/2018/5706142. eCollection 2018. J Ophthalmol. 2018. PMID: 29854428 Free PMC article.
-
Clinical and genetic features of eight Chinese autosomal-dominant optic atrophy pedigrees with six novel OPA1 pathogenic variants.Ophthalmic Genet. 2018 Oct;39(5):569-576. doi: 10.1080/13816810.2018.1466337. Epub 2018 Jun 28. Ophthalmic Genet. 2018. PMID: 29952689 Free PMC article.
References
-
- Lyle WM. Genetic risks: a reference for eye care practitioners. Waterloo (Canada): University of Waterloo Press; 1990.
-
- Eiberg H, Kjer B, Kjer P, Rosenberg T. Dominant optic atrophy (OPA1) mapped to chromosome 3q region I. Linkage analysis. Hum Mol Genet. 1994;3:977–80. - PubMed
-
- Kjer B, Eiberg H, Kjer P, Rosenberg T. Dominant optic atrophy mapped to chromosome 3q region. II. Clinical and epidemiological aspects. Acta Ophthalmol Scand. 1996;74:3–7. - PubMed
-
- Thiselton DL, Alexander C, Morris A, Brooks S, Rosenberg T, Eiberg H, Kjer B, Kjer P, Bhattacharya SS, Votruba M. A frameshift mutation in exon 28 of the OPA1 gene explains the high prevalence of dominant optic atrophy in the Danish population: evidence for a founder effect. Hum Genet. 2001;109:498–502. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
