

Neuroinflammation and oxidative stress in diabetic neuropathy: futuristic strategies based on these targets
- PMID: 24883061
- PMCID: PMC4021687
- DOI: 10.1155/2014/674987
Neuroinflammation and oxidative stress in diabetic neuropathy: futuristic strategies based on these targets
Abstract
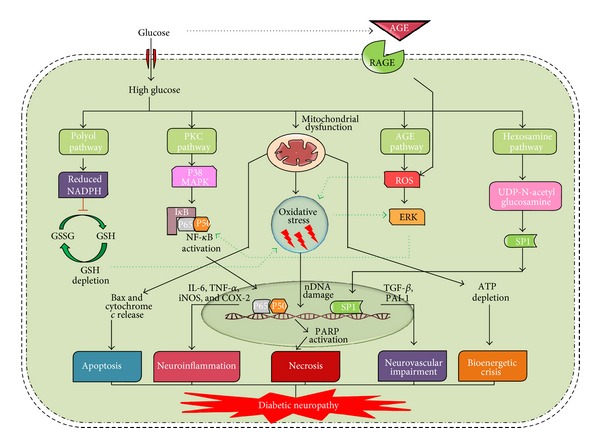
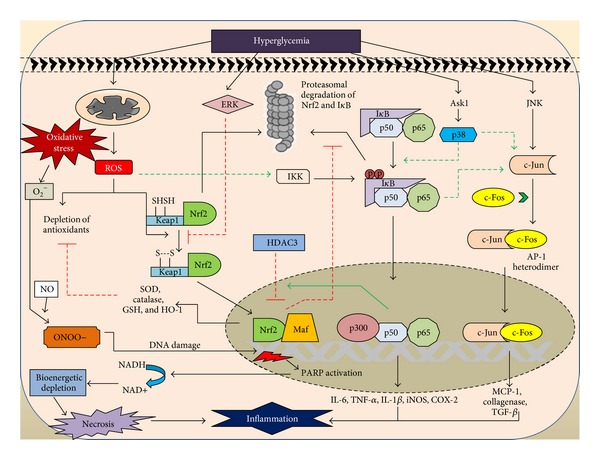
In Diabetes, the chronic hyperglycemia and associated complications affecting peripheral nerves are one of the most commonly occurring microvascular complications with an overall prevalence of 50-60%. Among the vascular complications of diabetes, diabetic neuropathy is the most painful and disabling, fatal complication affecting the quality of life in patients. Several theories of etiologies surfaced down the lane, amongst which the oxidative stress mediated damage in neurons and surrounding glial cell has gained attention as one of the vital mechanisms in the pathogenesis of neuropathy. Mitochondria induced ROS and other oxidants are responsible for altering the balance between oxidants and innate antioxidant defence of the body. Oxidative-nitrosative stress not only activates the major pathways namely, polyol pathway flux, advanced glycation end products formation, activation of protein kinase C, and overactivity of the hexosamine pathway, but also initiates and amplifies neuroinflammation. The cross talk between oxidative stress and inflammation is due to the activation of NF- κ B and AP-1 and inhibition of Nrf2, peroxynitrite mediate endothelial dysfunction, altered NO levels, and macrophage migration. These all culminate in the production of proinflammatory cytokines which are responsible for nerve tissue damage and debilitating neuropathies. This review focuses on the relationship between oxidative stress and neuroinflammation in the development and progression of diabetic neuropathy.
Figures
Similar articles
-
The Role of Oxidative Stress in Diabetic Neuropathy: Generation of Free Radical Species in the Glycation Reaction and Gene Polymorphisms Encoding Antioxidant Enzymes to Genetic Susceptibility to Diabetic Neuropathy in Population of Type I Diabetic Patients.Cell Biochem Biophys. 2015 Apr;71(3):1425-43. doi: 10.1007/s12013-014-0365-y. Cell Biochem Biophys. 2015. PMID: 25427889
-
A Review on Cellular and Molecular Mechanisms Linked to the Development of Diabetes Complications.Curr Diabetes Rev. 2021;17(4):457-473. doi: 10.2174/1573399816666201103143818. Curr Diabetes Rev. 2021. PMID: 33143626 Review.
-
Oxidative stress and diabetic retinopathy: Molecular mechanisms, pathogenetic role and therapeutic implications.Redox Biol. 2020 Oct;37:101799. doi: 10.1016/j.redox.2020.101799. Epub 2020 Nov 13. Redox Biol. 2020. PMID: 33248932 Free PMC article. Review.
-
Oxidative Stress in Diabetic Peripheral Neuropathy: Pathway and Mechanism-Based Treatment.Mol Neurobiol. 2023 Aug;60(8):4574-4594. doi: 10.1007/s12035-023-03342-7. Epub 2023 Apr 28. Mol Neurobiol. 2023. PMID: 37115404 Review.
-
Glucotoxic Mechanisms and Related Therapeutic Approaches.Int Rev Neurobiol. 2016;127:121-49. doi: 10.1016/bs.irn.2016.03.006. Epub 2016 Apr 8. Int Rev Neurobiol. 2016. PMID: 27133148 Review.
Cited by
-
Effects of Azadirachta indica on neuropathic pain induced by chronic constriction injury to sciatic nerve of Wistar rat.J Adv Vet Anim Res. 2022 Sep 29;9(3):359-368. doi: 10.5455/javar.2022.i603. eCollection 2022 Sep. J Adv Vet Anim Res. 2022. PMID: 36382046 Free PMC article.
-
Acetyllevocarnitine Hydrochloride for the Treatment of Diabetic Peripheral Neuropathy: A Phase 3 Randomized Clinical Trial in China.Diabetes. 2024 May 1;73(5):797-805. doi: 10.2337/db23-0377. Diabetes. 2024. PMID: 38320260 Free PMC article. Clinical Trial.
-
Neuroprotective effects of vitamin D on high fat diet- and palmitic acid-induced enteric neuronal loss in mice.BMC Gastroenterol. 2018 Nov 21;18(1):175. doi: 10.1186/s12876-018-0905-9. BMC Gastroenterol. 2018. PMID: 30463517 Free PMC article.
-
Effective fraction of Bletilla striata reduces the inflammatory cytokine production induced by water and organic extracts of airborne fine particulate matter (PM2.5) in vitro.BMC Complement Altern Med. 2019 Dec 16;19(1):369. doi: 10.1186/s12906-019-2790-3. BMC Complement Altern Med. 2019. PMID: 31842843 Free PMC article.
-
PGE2, Kidney Disease, and Cardiovascular Risk: Beyond Hypertension and Diabetes.J Am Soc Nephrol. 2016 Mar;27(3):666-76. doi: 10.1681/ASN.2015050528. Epub 2015 Aug 28. J Am Soc Nephrol. 2016. PMID: 26319242 Free PMC article. Review.
References
-
- IDF Diabetes Atlas. 6th edition. International Diabetes Federation; 2013.
-
- IDF Diabetes Atlas. 5th edition. International Diabetes Federation; 2012.
-
- Huizinga MM, Peltier A. Painful diabetic neuropathy: a management-centered review. Clinical Diabetes. 2007;25(1):6–15.
-
- Vinik AI, Mehrabyan A. Diabetic neuropathies. Medical Clinics of North America. 2004;88(4):947–999. - PubMed
-
- Oates PJ. Aldose reductase, still a compelling target for diabetic neuropathy. Current Drug Targets. 2008;9(1):14–36. - PubMed
Publication types
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
