

Hereditary breast and ovarian cancer: assessment of point mutations and copy number variations in Brazilian patients
- PMID: 24884479
- PMCID: PMC4038072
- DOI: 10.1186/1471-2350-15-55
Hereditary breast and ovarian cancer: assessment of point mutations and copy number variations in Brazilian patients
Abstract
Background: Germ line mutations in BRCA1 and BRCA2 (BRCA1/2) and other susceptibility genes have been identified as genetic causes of hereditary breast and ovarian cancer (HBOC). To identify the disease-causing mutations in a cohort of 120 Brazilian women fulfilling criteria for HBOC, we carried out a comprehensive screening of BRCA1/2, TP53 R337H, CHEK2 1100delC, followed by an analysis of copy number variations in 14 additional breast cancer susceptibility genes (PTEN, ATM, NBN, RAD50, RAD51, BRIP1, PALB2, MLH1, MSH2, MSH6, TP53, CDKN2A, CDH1 and CTNNB1).
Methods: Capillary sequencing and multiplex ligation-dependent probe amplification (MLPA) were used for detecting point mutations and copy number variations (CNVs), respectively, for the BRCA1 and BRCA2 genes; capillary sequencing was used for point mutation for both variants TP53 R337H and CHEK2 1100delC, and finally array comparative genomic hybridization (array-CGH) was used for identifying CNVs in the 14 additional genes.
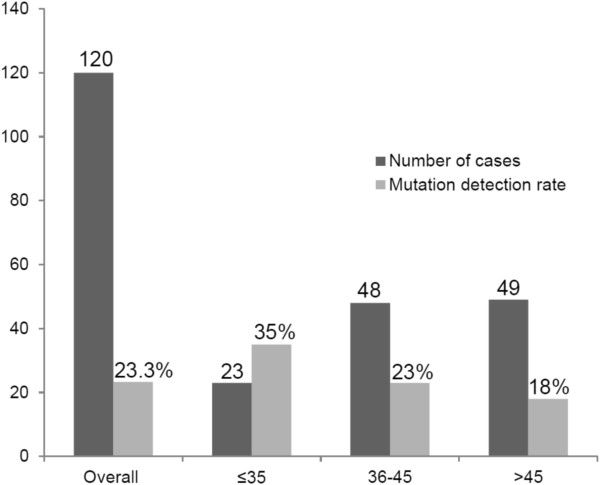
Results: The positive detection rate in our series was 26%. BRCA1 pathogenic mutations were found in 20 cases, including two cases with CNVs, whereas BRCA2 mutations were found in 7 cases. We also found three patients with the TP53 R337H mutation and one patient with the CHEK2 1100delC mutation. Seven (25%) pathogenic mutations in BRCA1/2 were firstly described, including a splice-site BRCA1 mutation for which pathogenicity was confirmed by the presence of an aberrant transcript showing the loss of the last 62 bp of exon 7. Microdeletions of exon 4 in ATM and exon 2 in PTEN were identified in BRCA2-mutated and BRCA1/2-negative patients, respectively.
Conclusions: In summary, our results showed a high frequency of BRCA1/2 mutations and a higher prevalence of BRCA1 (64.5%) gene. Moreover, the detection of the TP53 R337H variant in our series and the fact that this variant has a founder effect in our population prompted us to suggest that all female breast cancer patients with clinical criteria for HBOC and negative for BRCA1/2 genes should be tested for the TP53 R337H variant. Furthermore, the presence of genomic structural rearrangement resulting in CNVs in other genes that predispose breast cancer in conjunction with BRCA2 point mutations demonstrated a highly complex genetic etiology in Brazilian breast cancer families.
Figures



Similar articles
-
Screening for BRCA1, BRCA2, CHEK2, PALB2, BRIP1, RAD50, and CDH1 mutations in high-risk Finnish BRCA1/2-founder mutation-negative breast and/or ovarian cancer individuals.Breast Cancer Res. 2011 Feb 28;13(1):R20. doi: 10.1186/bcr2832. Breast Cancer Res. 2011. PMID: 21356067 Free PMC article.
-
Germline variants in DNA repair genes associated with hereditary breast and ovarian cancer syndrome: analysis of a 21 gene panel in the Brazilian population.BMC Med Genomics. 2020 Feb 10;13(1):21. doi: 10.1186/s12920-019-0652-y. BMC Med Genomics. 2020. PMID: 32039725 Free PMC article.
-
Mutation screening of TP53, CHEK2 and BRCA genes in patients at high risk for hereditary breast and ovarian cancer (HBOC) in Brazil.Breast Cancer. 2019 May;26(3):397-405. doi: 10.1007/s12282-018-00938-z. Epub 2018 Dec 11. Breast Cancer. 2019. PMID: 30535581
-
Systematic review of the molecular basis of hereditary breast and ovarian cancer syndrome in Brazil: the current scenario.Eur J Med Res. 2024 Mar 20;29(1):187. doi: 10.1186/s40001-024-01767-x. Eur J Med Res. 2024. PMID: 38504328 Free PMC article.
-
Pathogenic variants in BRCA1 and BRCA2 genes associated with female breast and ovarian cancer in the Mexican population.J Med Life. 2025 Jan;18(1):38-47. doi: 10.25122/jml-2024-0213. J Med Life. 2025. PMID: 40071159 Free PMC article.
Cited by
-
Familial history and prevalence of BRCA1, BRCA2 and TP53 pathogenic variants in HBOC Brazilian patients from a public healthcare service.Sci Rep. 2022 Nov 3;12(1):18629. doi: 10.1038/s41598-022-23012-3. Sci Rep. 2022. PMID: 36329109 Free PMC article.
-
Co-Occurrence of Germline Genomic Variants and Copy Number Variations in Hereditary Breast and Colorectal Cancer Patients.Genes (Basel). 2023 Aug 3;14(8):1580. doi: 10.3390/genes14081580. Genes (Basel). 2023. PMID: 37628631 Free PMC article.
-
TP53 p.R337H Germline Variant among Women at Risk of Hereditary Breast Cancer in a Public Health System of Midwest Brazil.Genes (Basel). 2024 Jul 16;15(7):928. doi: 10.3390/genes15070928. Genes (Basel). 2024. PMID: 39062707 Free PMC article.
-
Germline molecular data in hereditary breast cancer in Brazil: Lessons from a large single-center analysis.PLoS One. 2021 Feb 19;16(2):e0247363. doi: 10.1371/journal.pone.0247363. eCollection 2021. PLoS One. 2021. PMID: 33606809 Free PMC article.
-
The Current State of Breast Cancer Genetics in Populations of African Ancestry.Genes (Basel). 2025 Feb 6;16(2):199. doi: 10.3390/genes16020199. Genes (Basel). 2025. PMID: 40004528 Free PMC article. Review.
References
-
- Ford D, Easton DF, Stratton M, Narod S, Goldgar D, Devilee P, Bishop DT, Weber B, Lenoir G, Chang-Claude J, Sobol H, Teare MD, Struewing J, Arason A, Scherneck S, Peto J, Rebbeck TR, Tonin P, Neuhausen S, Barkardottir R, Eyfjord J, Lynch H, Ponder BA, Gayther SA, Zelada-Hedman M. and the Breast Cancer Linkage Consortium. Genetic heterogeneity and penetrance analysis of the BRCA1 and BRCA2 genes in breast cancer families. the Breast Cancer Linkage Consortium. Am J Hum Genet. 1998;15(3):676–689. doi: 10.1086/301749. - DOI - PMC - PubMed
-
- Thorlacius S, Olafsdottir G, Tryggvadottir L, Neuhausen S, Jonasson JG, Tavtigian SV, Tulinius H, Ogmundsdottir HM, Eyfjörd JE. A single BRCA2 mutation in male and female breast cancer families from Iceland with varied cancer phenotypes. Nat Genet. 1996;15(1):117–119. doi: 10.1038/ng0596-117. - DOI - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous
