

Direct squencing from the minimal number of DNA molecules needed to fill a 454 picotiterplate
- PMID: 24887077
- PMCID: PMC4041646
- DOI: 10.1371/journal.pone.0097379
Direct squencing from the minimal number of DNA molecules needed to fill a 454 picotiterplate
Erratum in
- PLoS One. 2014;9(7):e102719
Abstract
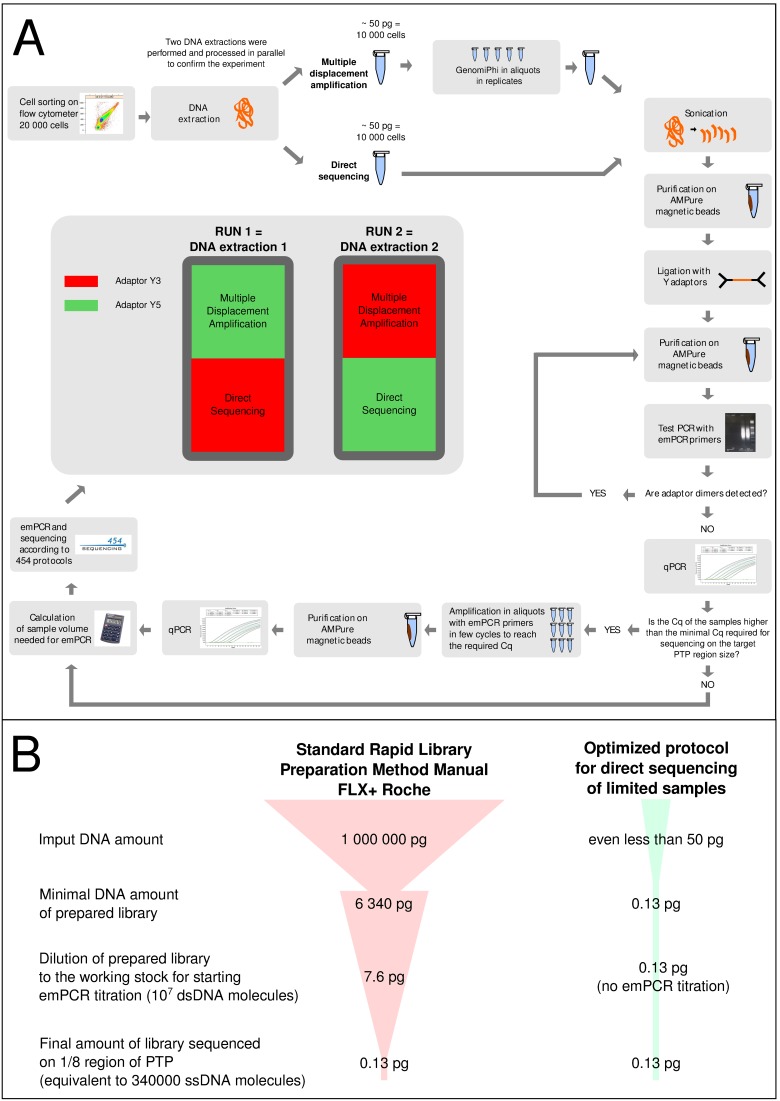
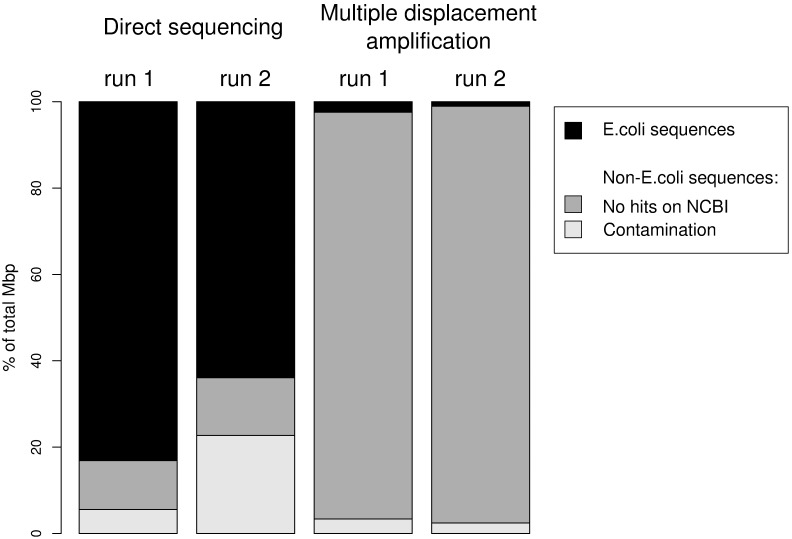
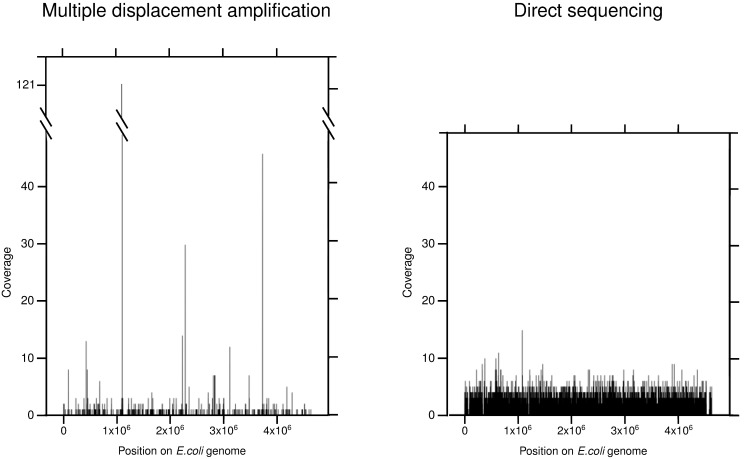
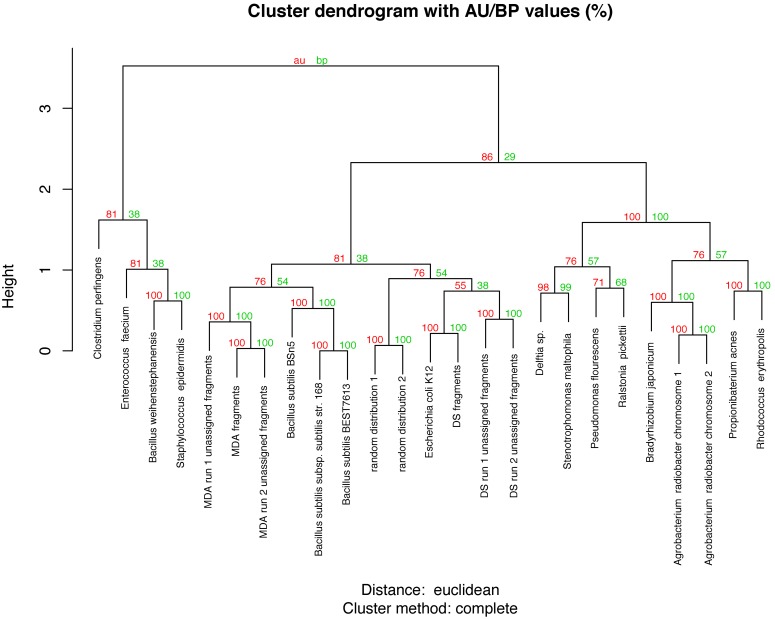
The large amount of DNA needed to prepare a library in next generation sequencing protocols hinders direct sequencing of small DNA samples. This limitation is usually overcome by the enrichment of such samples with whole genome amplification (WGA), mostly by multiple displacement amplification (MDA) based on φ29 polymerase. However, this technique can be biased by the GC content of the sample and is prone to the development of chimeras as well as contamination during enrichment, which contributes to undesired noise during sequence data analysis, and also hampers the proper functional and/or taxonomic assignments. An alternative to MDA is direct DNA sequencing (DS), which represents the theoretical gold standard in genome sequencing. In this work, we explore the possibility of sequencing the genome of Escherichia coli fs 24 from the minimum number of DNA molecules required for pyrosequencing, according to the notion of one-bead-one-molecule. Using an optimized protocol for DS, we constructed a shotgun library containing the minimum number of DNA molecules needed to fill a selected region of a picotiterplate. We gathered most of the reference genome extension with uniform coverage. We compared the DS method with MDA applied to the same amount of starting DNA. As expected, MDA yielded a sparse and biased read distribution, with a very high amount of unassigned and unspecific DNA amplifications. The optimized DS protocol allows unbiased sequencing to be performed from samples with a very small amount of DNA.
Conflict of interest statement
Figures
Similar articles
-
Assessment of REPLI-g Multiple Displacement Whole Genome Amplification (WGA) Techniques for Metagenomic Applications.J Biomol Tech. 2017 Apr;28(1):46-55. doi: 10.7171/jbt.17-2801-008. Epub 2017 Mar 21. J Biomol Tech. 2017. PMID: 28344519 Free PMC article.
-
Systematic Characteristic Exploration of the Chimeras Generated in Multiple Displacement Amplification through Next Generation Sequencing Data Reanalysis.PLoS One. 2015 Oct 6;10(10):e0139857. doi: 10.1371/journal.pone.0139857. eCollection 2015. PLoS One. 2015. PMID: 26440104 Free PMC article.
-
Optimized Workflow for Whole Genome and Transcriptome Next-Generation Sequencing of Single Cells or Limited Nucleic Acid Samples.Curr Protoc. 2023 May;3(5):e753. doi: 10.1002/cpz1.753. Curr Protoc. 2023. PMID: 37166214
-
Whole Genome Library Construction for Next Generation Sequencing.Methods Mol Biol. 2018;1706:151-161. doi: 10.1007/978-1-4939-7471-9_8. Methods Mol Biol. 2018. PMID: 29423797 Review.
-
Whole genome amplification with Phi29 DNA polymerase to enable genetic or genomic analysis of samples of low DNA yield.Methods Mol Biol. 2008;439:1-18. doi: 10.1007/978-1-59745-188-8_1. Methods Mol Biol. 2008. PMID: 18370092 Review.
Cited by
-
Exploring the human microbiome from multiple perspectives: factors altering its composition and function.FEMS Microbiol Rev. 2017 Jul 1;41(4):453-478. doi: 10.1093/femsre/fuw046. FEMS Microbiol Rev. 2017. PMID: 28333226 Free PMC article. Review.
-
Expanding a Wastewater-Based Surveillance Methodology for DNA Isolation from a Workflow Optimized for SARS-CoV-2 RNA Quantification.J Biomol Tech. 2023 Dec 20;34(4):3fc1f5fe.dfa8d906. doi: 10.7171/3fc1f5fe.dfa8d906. eCollection 2023 Dec. J Biomol Tech. 2023. PMID: 38268997 Free PMC article.
-
CleanBar: a versatile demultiplexing tool for split-and-pool barcoding in single-cell omics.ISME Commun. 2025 Aug 1;5(1):ycaf134. doi: 10.1093/ismeco/ycaf134. eCollection 2025 Jan. ISME Commun. 2025. PMID: 40860566 Free PMC article.
-
Metagenomic assessment of the interplay between the environment and the genetic diversification of Acinetobacter.Environ Microbiol. 2017 Dec;19(12):5010-5024. doi: 10.1111/1462-2920.13949. Epub 2017 Dec 1. Environ Microbiol. 2017. PMID: 28967182 Free PMC article.
-
A simple, reproducible and cost-effective procedure to analyse gut phageome: from phage isolation to bioinformatic approach.Sci Rep. 2019 Aug 5;9(1):11331. doi: 10.1038/s41598-019-47656-w. Sci Rep. 2019. PMID: 31383878 Free PMC article.
References
-
- Binga EK, Lasken RS, Neufeld JD (2008) Something from (almost) nothing: the impact of multiple displacement amplification on microbial ecology. ISME J 2: 233–241. - PubMed
-
- Vlček Č, Pačes V (1986) Nucleotide sequence of the late region of bacillus phage phi 29 completes the 19285-bp sequence of phi 29 genome. Comparison with the homologous sequence of phage PZA. Gene 46: 215–225. - PubMed
Publication types
MeSH terms
Associated data
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
