

Ginsenoside Rh1 potentiates dexamethasone's anti-inflammatory effects for chronic inflammatory disease by reversing dexamethasone-induced resistance
- PMID: 24887434
- PMCID: PMC4060561
- DOI: 10.1186/ar4556
Ginsenoside Rh1 potentiates dexamethasone's anti-inflammatory effects for chronic inflammatory disease by reversing dexamethasone-induced resistance
Abstract
Introduction: Acquired resistance to glucocorticoids constitutes a major clinical challenge, often overlooked in the search for compounds to improve the effect of classic steroids. We sought to unravel how a plant-original compound, ginsenoside Rh1, potentiates dexmethasone (DEX)'s potential anti-inflammation properties.
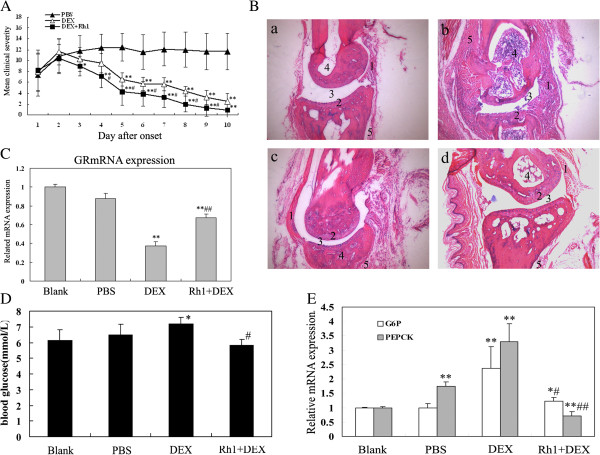
Methods: Ginsenoside Rh1 combined with DEX was applied in a short-term and long-term treatment protocol for inflammation. Its potential mechanism on anti-inflammation was explored. In addition, the effect of Rh1 on the side-effect induced by DEX was studied. Furthermore, the in vivo anti-inflammatory effects of Rh1 combined with DEX were evaluated in a collagen-induced arthritis (CIA) mice model.
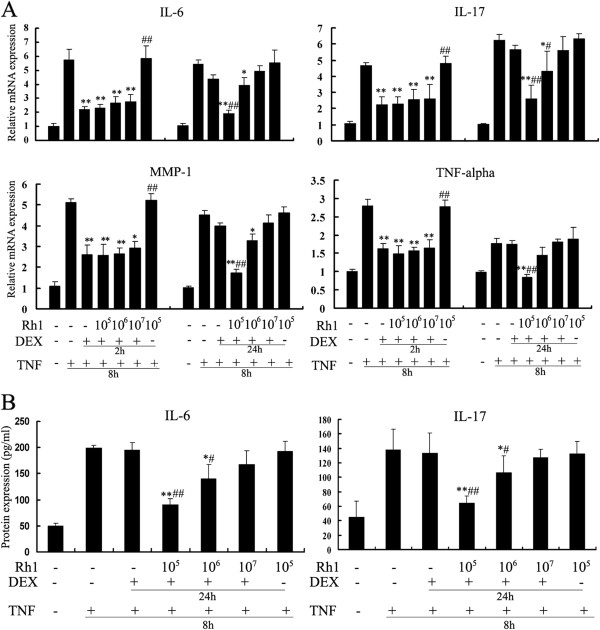
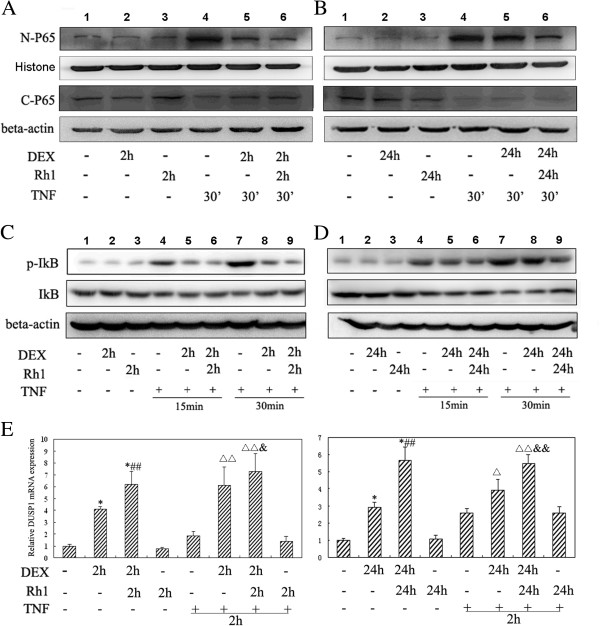
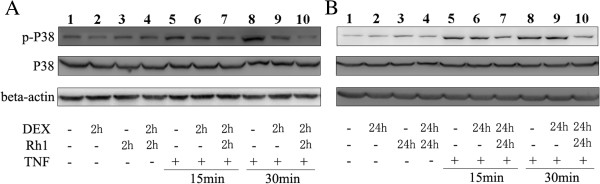
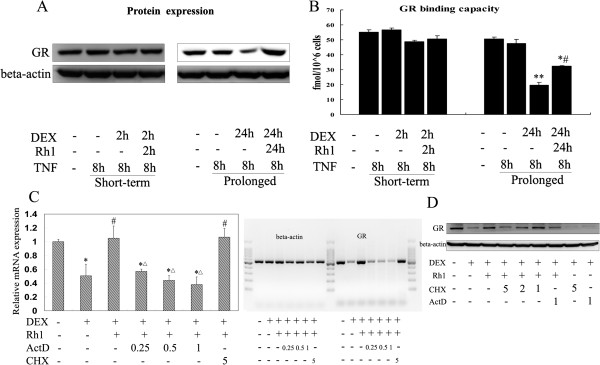
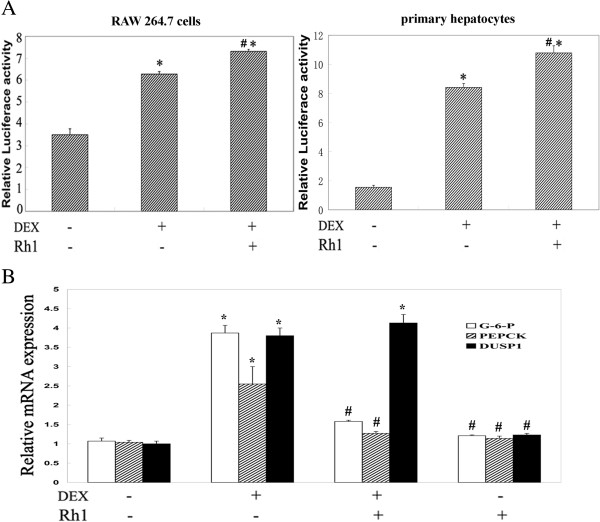
Results: Ginsenoside Rh1 potentiates DEX's anti-inflammatory effects even after prolonged DEX treatment. Rh1 could improve the glucocorticoid receptor (GR)'s transrepression on nuclear factor kappa B (NF-κB) and transactivation on dual specificity protein phosphatase 1 (DUSP1), which is responsible for DEX's anti-inflammatory effects. Parallel Western blot assay and radioligand binding analysis revealed that Rh1 could increase the expression and binding of GR. This is in sharp contrast to DEX alone, showing a direct link among prolonged treatment, decreasing GR and the abolishment of anti-inflammation. Interestingly, Rh1 does not enhance the transactivation of glucocorticoid-responsive elements (GRE) driven genes - gluconeogenic enzyme glucose-6-phosphatase (G6P) and phosphoenolpyruvate carboxykinasee phosphatase (PEPCK) in primary mouse hepatocytes, a mechanism partly held accountable for the metabolic side-effects. Similar results were found in CIA mice.
Conclusion: Rh1 could potentiate DEX's anti-inflammatory effects and does not cause a hyperglycemic side effect. Ginsenoside Rh1 combined with DEX may be a promising candidate treatment option for chronic inflammatory diseases in need of long-term immunosuppression therapies.
Figures
Similar articles
-
A plant-derived glucocorticoid receptor modulator attenuates inflammation without provoking ligand-induced resistance.Ann Rheum Dis. 2010 Jan;69(1):291-6. doi: 10.1136/ard.2008.102871. Ann Rheum Dis. 2010. PMID: 19204014
-
Minor Ginsenoside Rg2 and Rh1 Attenuates LPS-Induced Acute Liver and Kidney Damages via Downregulating Activation of TLR4-STAT1 and Inflammatory Cytokine Production in Macrophages.Int J Mol Sci. 2020 Sep 11;21(18):6656. doi: 10.3390/ijms21186656. Int J Mol Sci. 2020. PMID: 32932915 Free PMC article.
-
Anti-inflammatory effects of selective glucocorticoid receptor modulators are partially dependent on up-regulation of dual specificity phosphatase 1.Br J Pharmacol. 2012 Feb;165(4b):1124-36. doi: 10.1111/j.1476-5381.2011.01574.x. Br J Pharmacol. 2012. PMID: 21718312 Free PMC article.
-
Anti-inflammatory actions of glucocorticoids: molecular mechanisms.Clin Sci (Lond). 1998 Jun;94(6):557-72. doi: 10.1042/cs0940557. Clin Sci (Lond). 1998. PMID: 9854452 Review.
-
How glucocorticoid receptors modulate the activity of other transcription factors: a scope beyond tethering.Mol Cell Endocrinol. 2013 Nov 5;380(1-2):41-54. doi: 10.1016/j.mce.2012.12.014. Epub 2012 Dec 23. Mol Cell Endocrinol. 2013. PMID: 23267834 Review.
Cited by
-
Two new triterpenoid saponins derived from the leaves of Panax ginseng and their antiinflammatory activity.J Ginseng Res. 2019 Oct;43(4):600-605. doi: 10.1016/j.jgr.2018.09.004. Epub 2018 Oct 4. J Ginseng Res. 2019. PMID: 31695566 Free PMC article.
-
Disease- and treatment-associated acquired glucocorticoid resistance.Endocr Connect. 2018 Dec;7(12):R328-R349. doi: 10.1530/EC-18-0421. Endocr Connect. 2018. PMID: 30352419 Free PMC article. Review.
-
Novel role for receptor dimerization in post-translational processing and turnover of the GRα.Sci Rep. 2018 Sep 24;8(1):14266. doi: 10.1038/s41598-018-32440-z. Sci Rep. 2018. PMID: 30250038 Free PMC article.
-
Immunomodulatory, Anti-Inflammatory, and Anti-Cancer Properties of Ginseng: A Pharmacological Update.Molecules. 2023 May 3;28(9):3863. doi: 10.3390/molecules28093863. Molecules. 2023. PMID: 37175273 Free PMC article. Review.
-
The promising therapeutic potentials of ginsenosides mediated through p38 MAPK signaling inhibition.Heliyon. 2021 Nov 12;7(11):e08354. doi: 10.1016/j.heliyon.2021.e08354. eCollection 2021 Nov. Heliyon. 2021. PMID: 34825082 Free PMC article. Review.
References
-
- Sliwinska-Stanczyk P, Pazdur J, Ziolkowska M, Jaworski J, Kaminska-Tchorzewska E, Lacki JK. The effect of methylprednisolone on proliferation of PBMCs obtained from steroid-sensitive and steroid-resistant rheumatoid arthritis patients. Scand J Rheumatol. 2007;36:167–171. - PubMed
-
- Kirwan JR. Glucocorticoid resistance in patients with rheumatoid arthritis. Scand J Rheumatol. 2007;36:165–166. - PubMed
-
- Schaaf MJ, Cidlowski JA. Molecular mechanisms of glucocorticoid action and resistance. J Steroid Biochem Mol Biol. 2002;83:37–48. - PubMed
-
- Buttgereit F, Saag KG, Cutolo M, da Silva JA, Bijlsma JW. The molecular basis for the effectiveness, toxicity, and resistance to glucocorticoids: focus on the treatment of rheumatoid arthritis. Scand J Rheumatol. 2005;34:14–21. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
