

An integrated genomic approach identifies persistent tumor suppressive effects of transforming growth factor-β in human breast cancer
- PMID: 24890385
- PMCID: PMC4095608
- DOI: 10.1186/bcr3668
An integrated genomic approach identifies persistent tumor suppressive effects of transforming growth factor-β in human breast cancer
Abstract
Introduction: Transforming growth factor-βs (TGF-βs) play a dual role in breast cancer, with context-dependent tumor-suppressive or pro-oncogenic effects. TGF-β antagonists are showing promise in early-phase clinical oncology trials to neutralize the pro-oncogenic effects. However, there is currently no way to determine whether the tumor-suppressive effects of TGF-β are still active in human breast tumors at the time of surgery and treatment, a situation that could lead to adverse therapeutic responses.
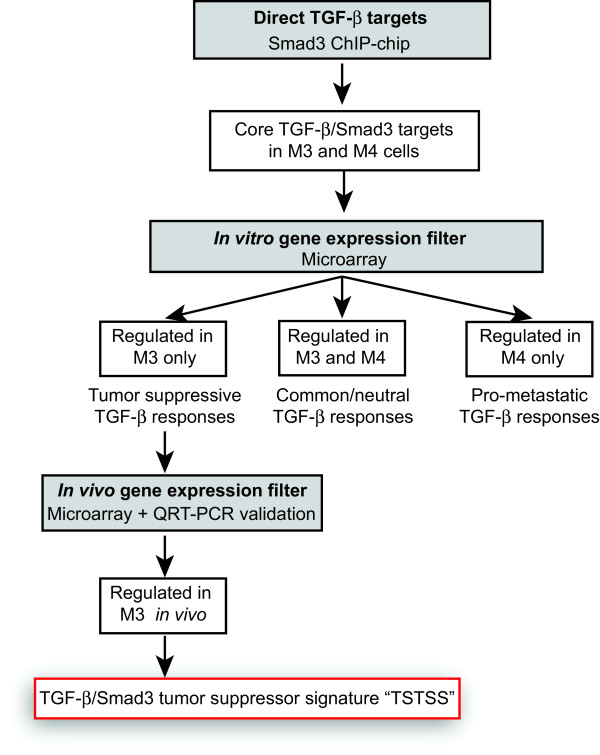
Methods: Using a breast cancer progression model that exemplifies the dual role of TGF-β, promoter-wide chromatin immunoprecipitation and transcriptomic approaches were applied to identify a core set of TGF-β-regulated genes that specifically reflect only the tumor-suppressor arm of the pathway. The clinical significance of this signature and the underlying biology were investigated using bioinformatic analyses in clinical breast cancer datasets, and knockdown validation approaches in tumor xenografts.
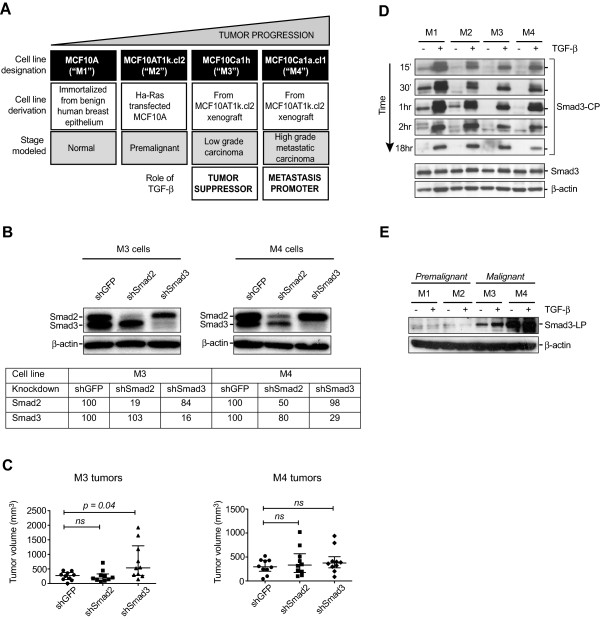
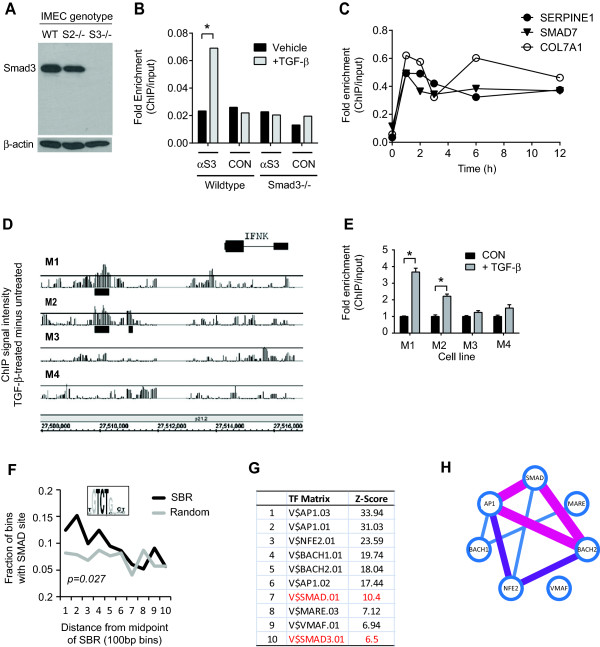
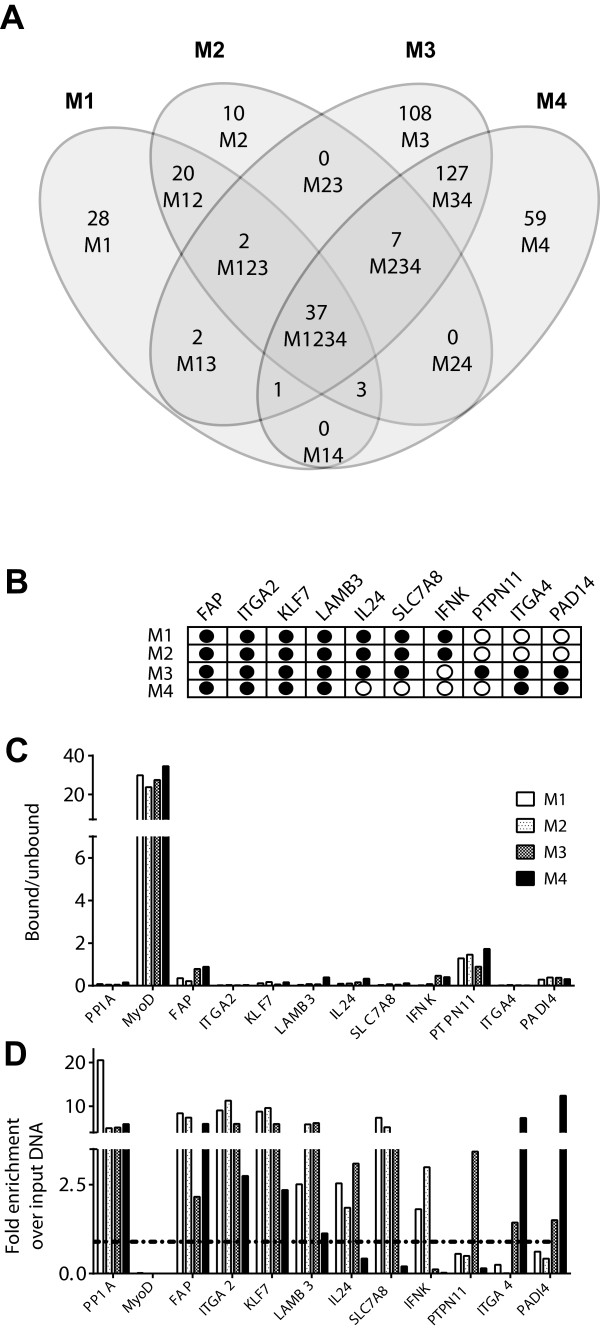
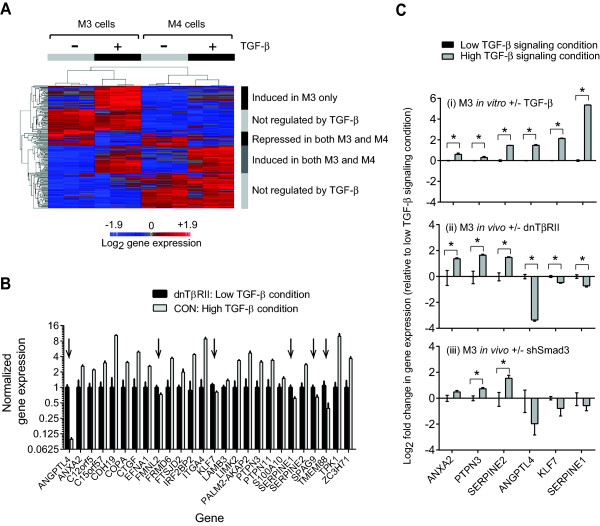
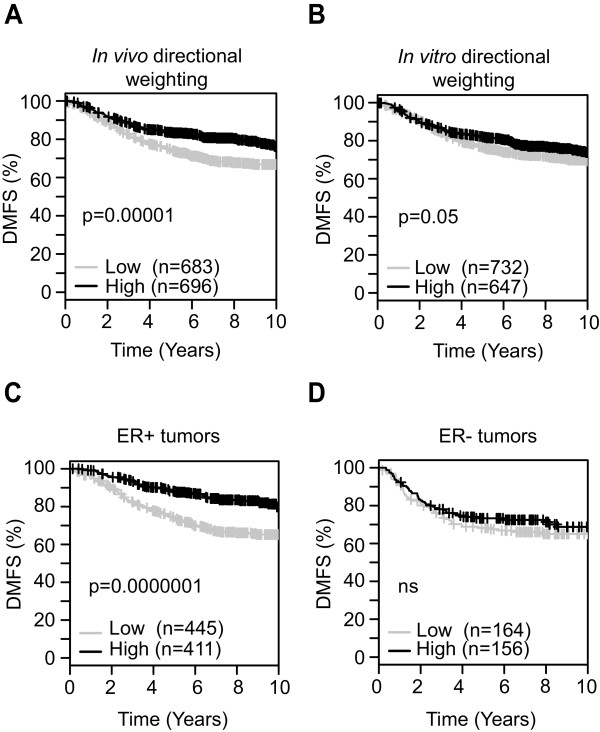
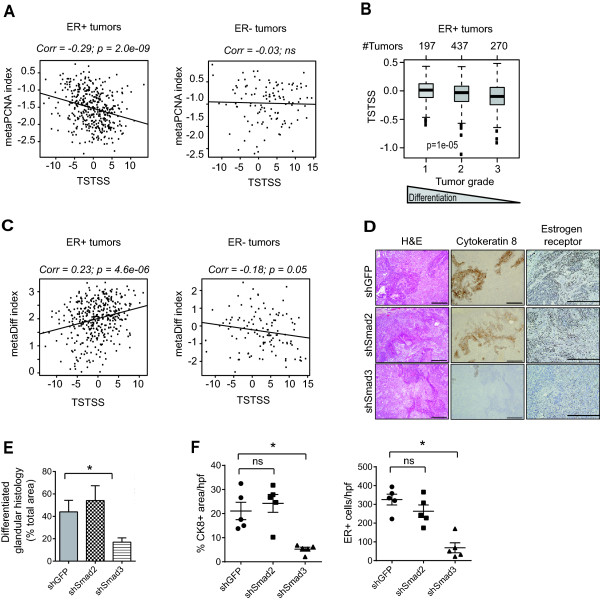
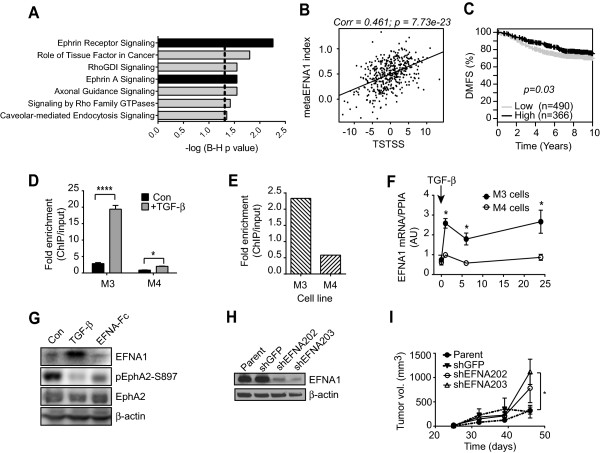
Results: TGF-β-driven tumor suppression was highly dependent on Smad3, and Smad3 target genes that were specifically enriched for involvement in tumor suppression were identified. Patterns of Smad3 binding reflected the preexisting active chromatin landscape, and target genes were frequently regulated in opposite directions in vitro and in vivo, highlighting the strong contextuality of TGF-β action. An in vivo-weighted TGF-β/Smad3 tumor-suppressor signature was associated with good outcome in estrogen receptor-positive breast cancer cohorts. TGF-β/Smad3 effects on cell proliferation, differentiation and ephrin signaling contributed to the observed tumor suppression.
Conclusions: Tumor-suppressive effects of TGF-β persist in some breast cancer patients at the time of surgery and affect clinical outcome. Carefully tailored in vitro/in vivo genomic approaches can identify such patients for exclusion from treatment with TGF-β antagonists.
Trial registration: ClinicalTrials.gov NCT01401062.
Figures
References
-
- Reis-Filho JS, Weigelt B, Fumagalli D, Sotiriou C. Molecular profiling: moving away from tumor philately. Sci Transl Med. 2010;2:ps43. - PubMed
-
- Ikushima H, Miyazono K. TGFβ signalling: a complex web in cancer progression. Nat Rev Cancer. 2010;10:415–424. - PubMed
-
- Pardali K, Moustakas A. Actions of TGF-β as tumor suppressor and pro-metastatic factor in human cancer. Biochim Biophys Acta. 2007;1775:21–62. - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
