

Epigenetic control of immunity
- PMID: 24890513
- PMCID: PMC4031963
- DOI: 10.1101/cshperspect.a019307
Epigenetic control of immunity
Abstract
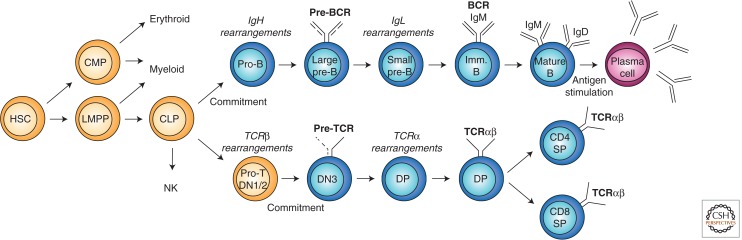
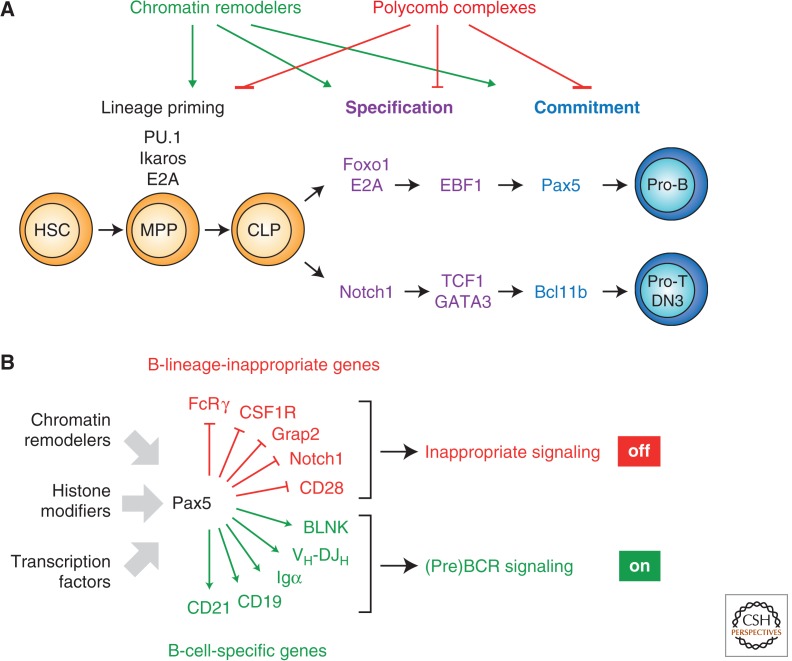
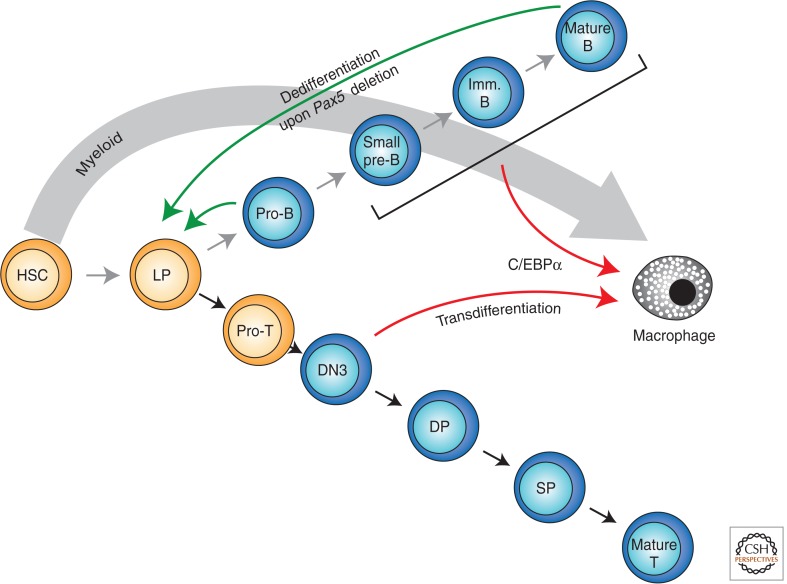
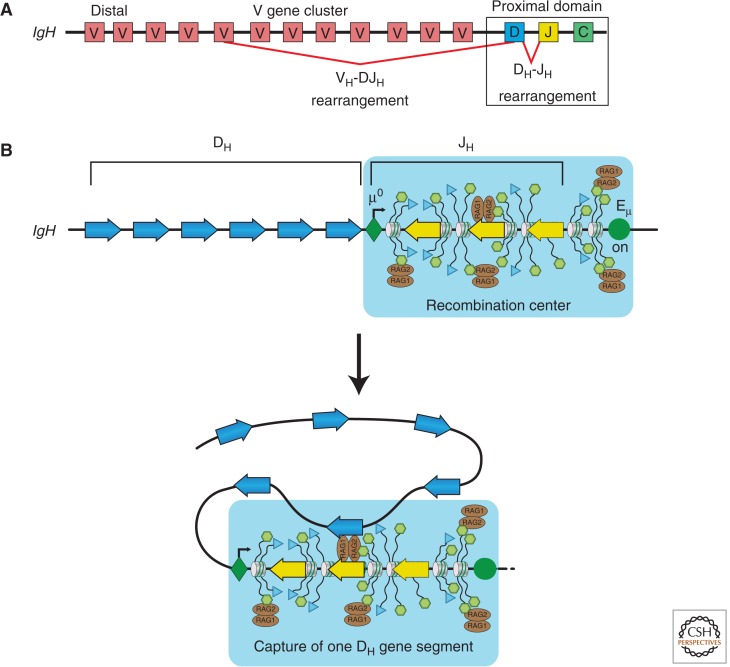
Immunity relies on the heterogeneity of immune cells and their ability to respond to pathogen challenges. In the adaptive immune system, lymphocytes display a highly diverse antigen receptor repertoire that matches the vast diversity of pathogens. In the innate immune system, the cell's heterogeneity and phenotypic plasticity enable flexible responses to changes in tissue homeostasis caused by infection or damage. The immune responses are calibrated by the graded activity of immune cells that can vary from yeast-like proliferation to lifetime dormancy. This article describes key epigenetic processes that contribute to the function of immune cells during health and disease.
Copyright © 2014 Cold Spring Harbor Laboratory Press; all rights reserved.
Figures
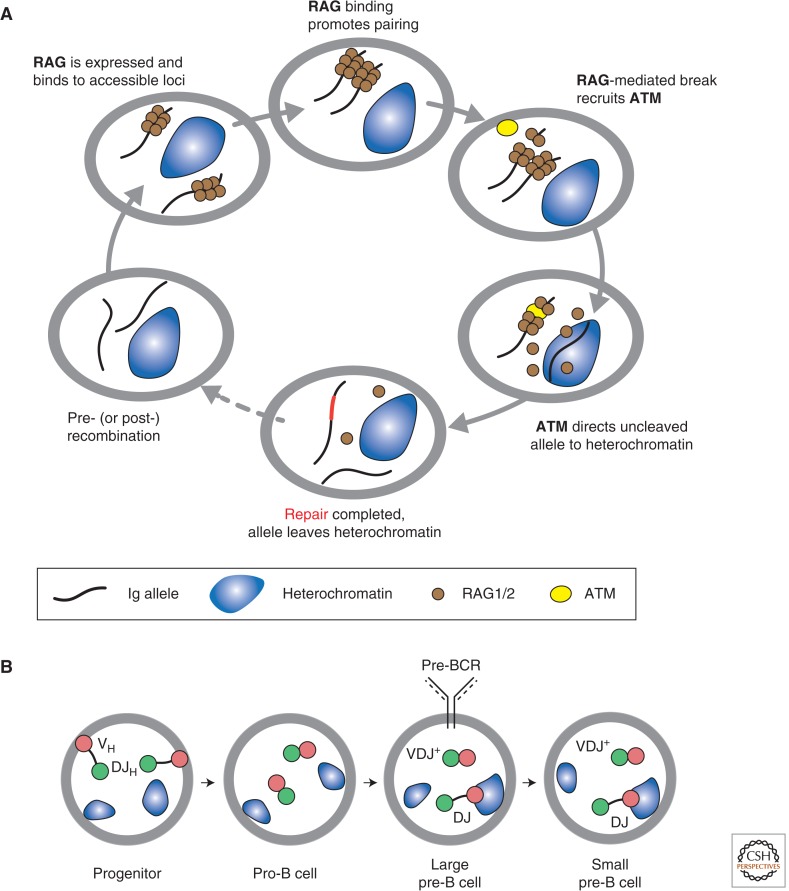
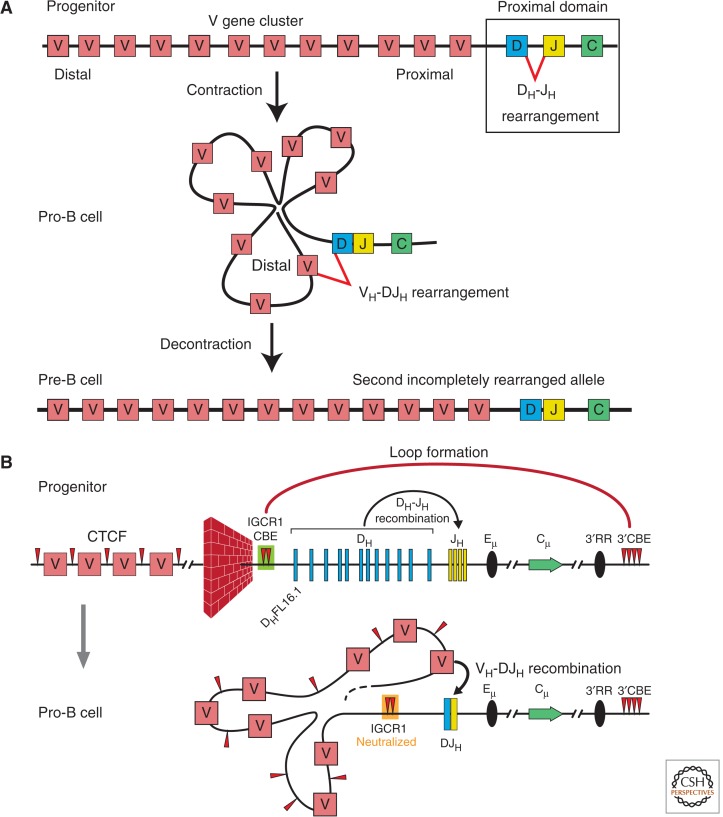
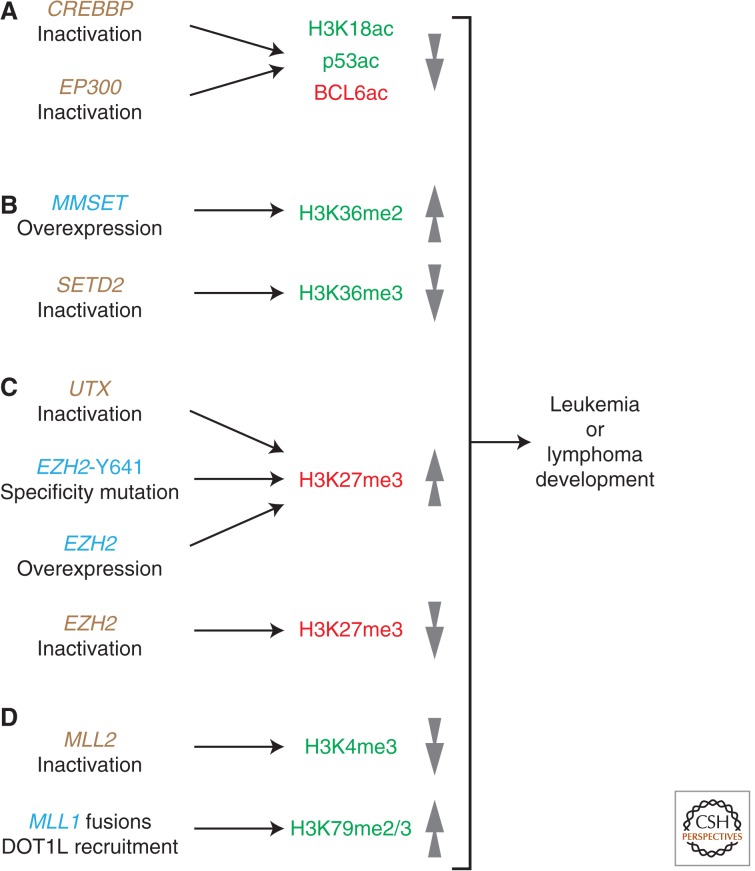
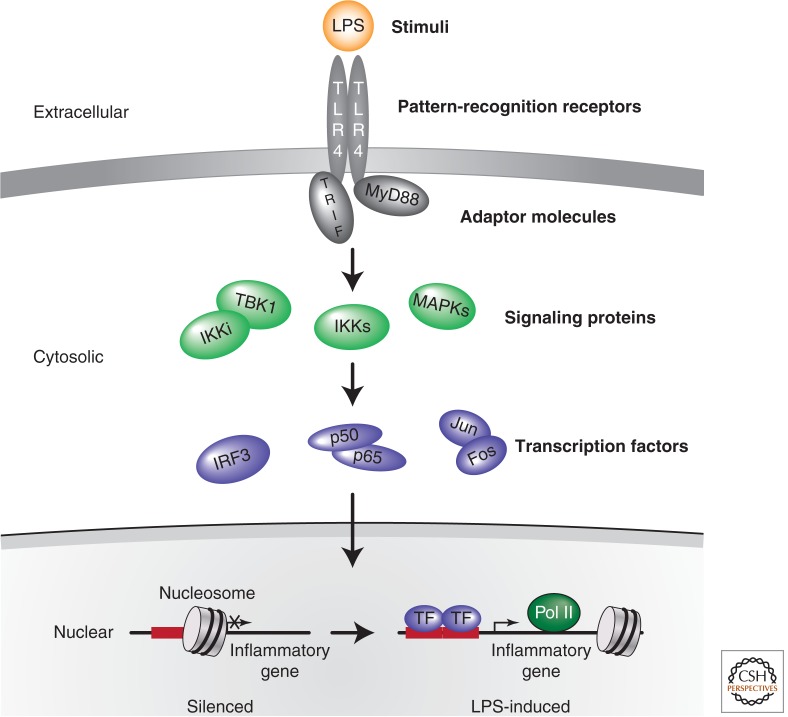
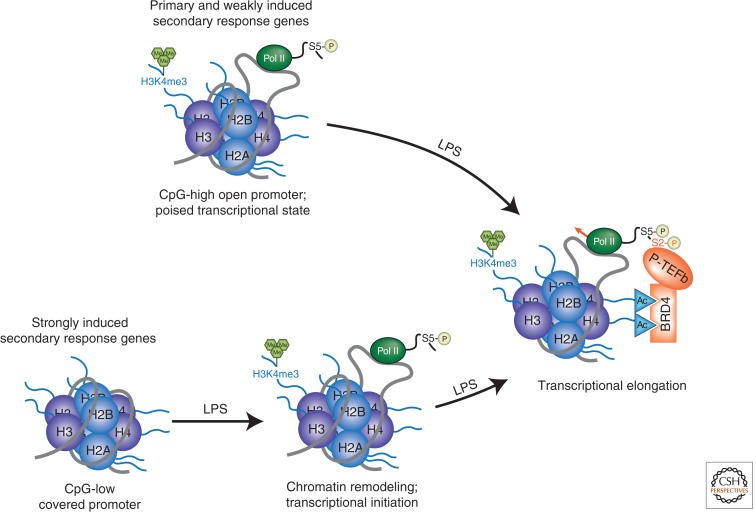
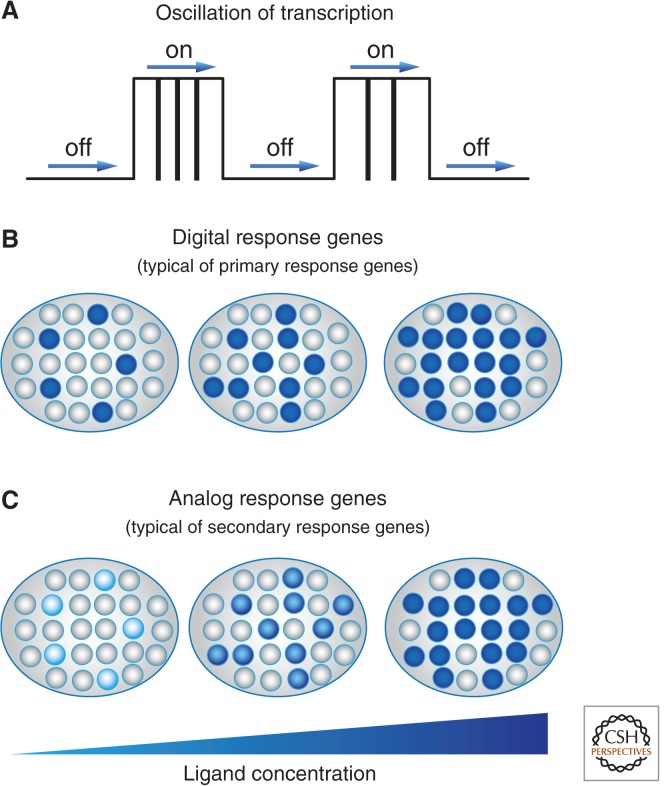
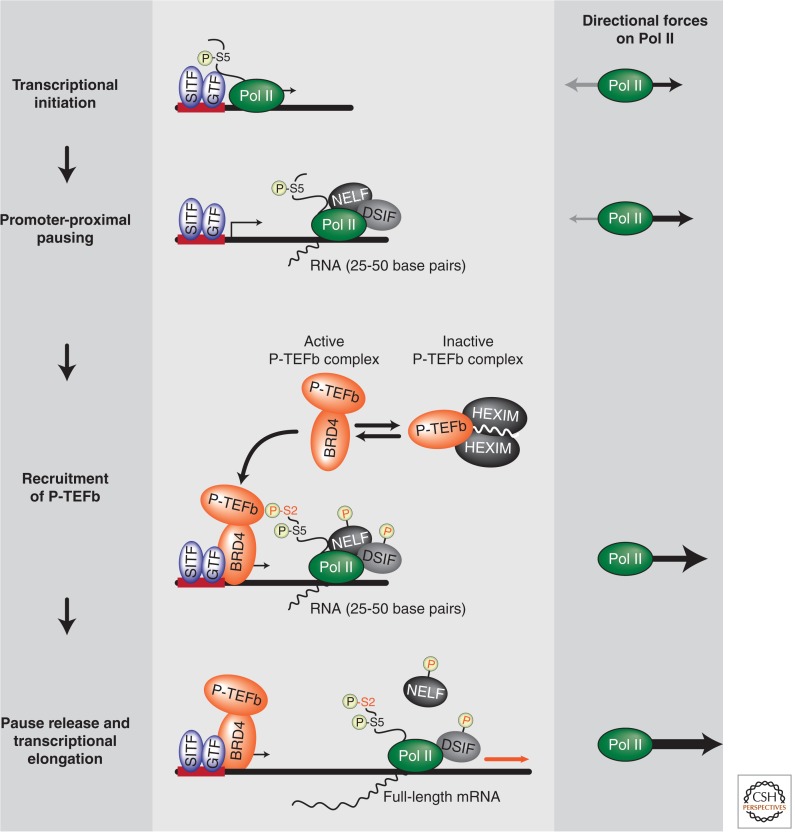
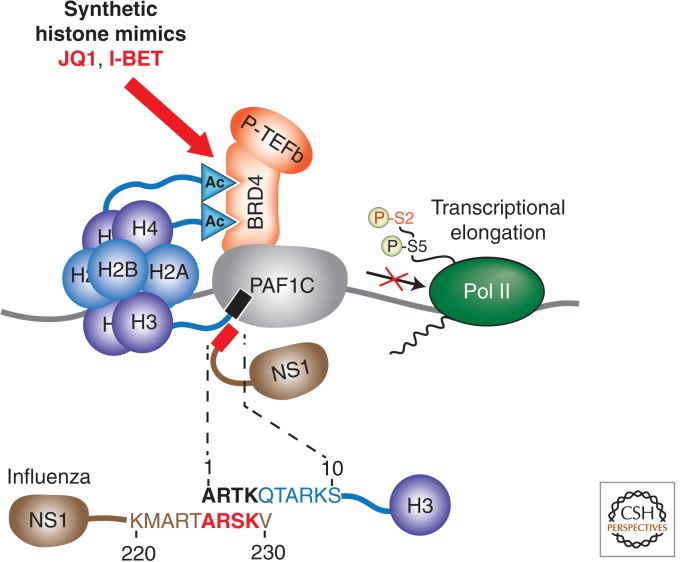
References
-
- Bacher CP, Guggiari M, Brors B, Augui S, Clerc P, Avner P, Eils R, Heard E 2006. Transient colocalization of X-inactivation centres accompanies the initiation of X inactivation. Nat Cell Biol 8: 293–299 - PubMed
-
- Bai L, Morozov AV 2010. Gene regulation by nucleosome positioning. Trends Genet 26: 476–483 - PubMed
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources