

Metabolite proofreading in carnosine and homocarnosine synthesis: molecular identification of PM20D2 as β-alanyl-lysine dipeptidase
- PMID: 24891507
- PMCID: PMC4094082
- DOI: 10.1074/jbc.M114.576579
Metabolite proofreading in carnosine and homocarnosine synthesis: molecular identification of PM20D2 as β-alanyl-lysine dipeptidase
Abstract
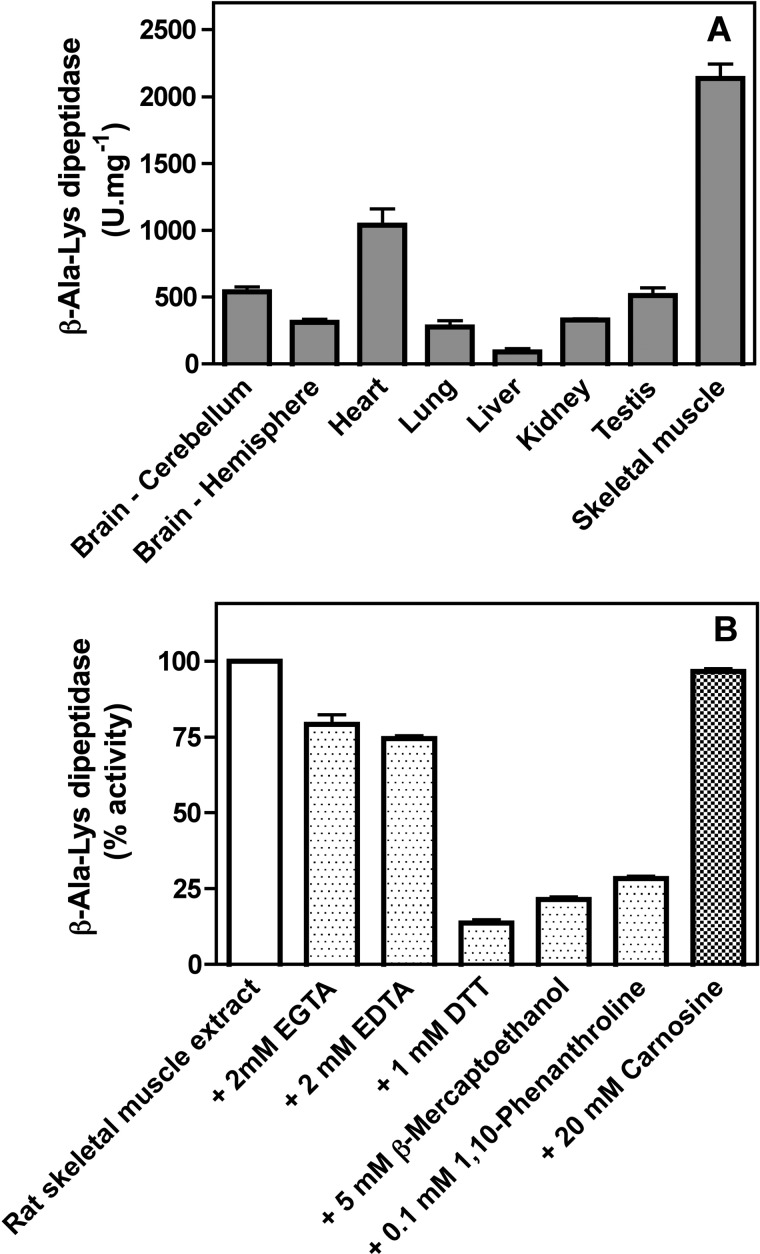
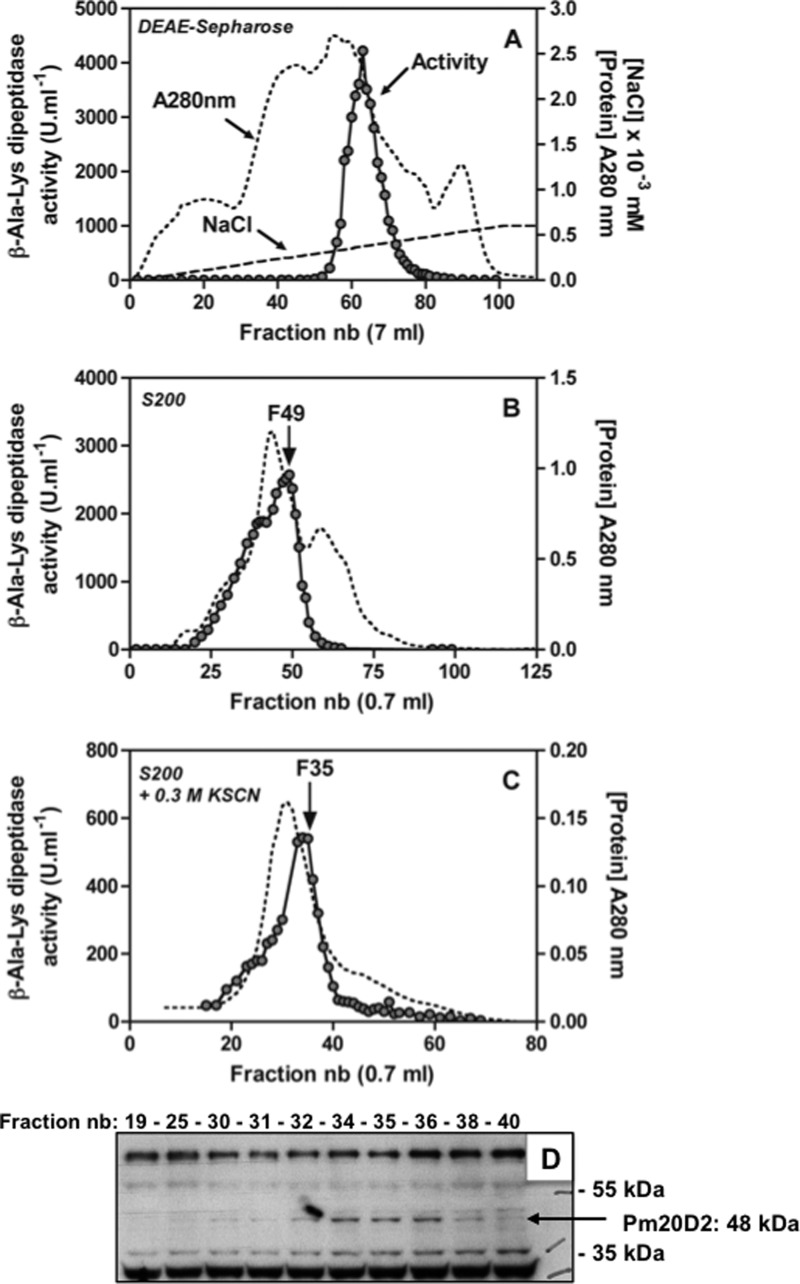
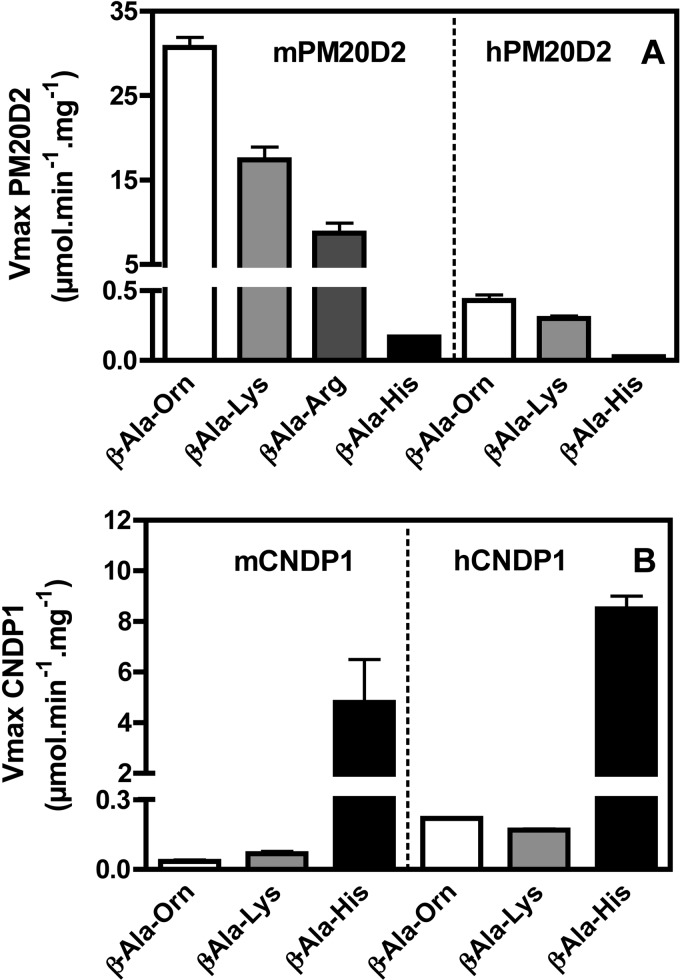
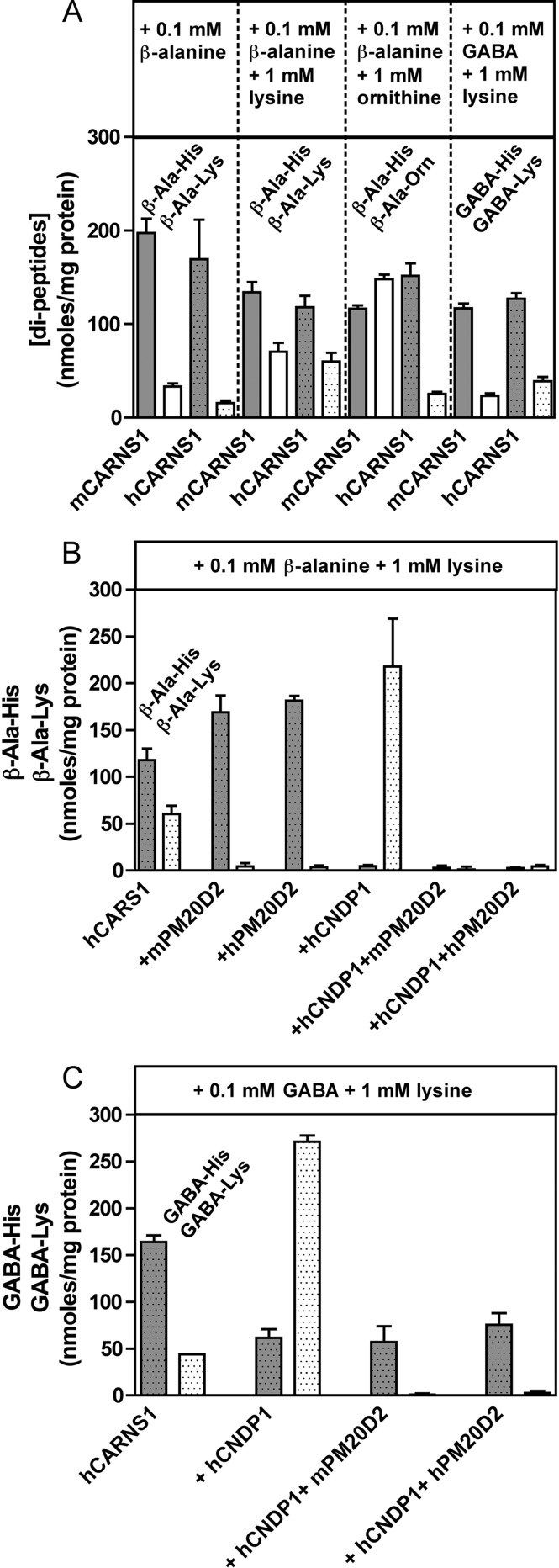
Carnosine synthase is the ATP-dependent ligase responsible for carnosine (β-alanyl-histidine) and homocarnosine (γ-aminobutyryl-histidine) synthesis in skeletal muscle and brain, respectively. This enzyme uses, also at substantial rates, lysine, ornithine, and arginine instead of histidine, yet the resulting dipeptides are virtually absent from muscle or brain, suggesting that they are removed by a "metabolite repair" enzyme. Using a radiolabeled substrate, we found that rat skeletal muscle, heart, and brain contained a cytosolic β-alanyl-lysine dipeptidase activity. This enzyme, which has the characteristics of a metalloenzyme, was purified ≈ 200-fold from rat skeletal muscle. Mass spectrometry analysis of the fractions obtained at different purification stages indicated parallel enrichment of PM20D2, a peptidase of unknown function belonging to the metallopeptidase 20 family. Western blotting showed coelution of PM20D2 with β-alanyl-lysine dipeptidase activity. Recombinant mouse PM20D2 hydrolyzed β-alanyl-lysine, β-alanyl-ornithine, γ-aminobutyryl-lysine, and γ-aminobutyryl-ornithine as its best substrates. It also acted at lower rates on β-alanyl-arginine and γ-aminobutyryl-arginine but virtually not on carnosine or homocarnosine. Although acting preferentially on basic dipeptides derived from β-alanine or γ-aminobutyrate, PM20D2 also acted at lower rates on some "classic dipeptides" like α-alanyl-lysine and α-lysyl-lysine. The same activity profile was observed with human PM20D2, yet this enzyme was ∼ 100-200-fold less active on all substrates tested than the mouse enzyme. Cotransfection in HEK293T cells of mouse or human PM20D2 together with carnosine synthase prevented the accumulation of abnormal dipeptides (β-alanyl-lysine, β-alanyl-ornithine, γ-aminobutyryl-lysine), thus favoring the synthesis of carnosine and homocarnosine and confirming the metabolite repair role of PM20D2.
Keywords: Brain Metabolism; CARNS1; CNDP1; Carnosine; Homocarnosine; M20 Metallopeptidase; Metabolism; Peptidase; Peptides; Skeletal Muscle Metabolism.
© 2014 by The American Society for Biochemistry and Molecular Biology, Inc.
Figures
References
-
- Linster C. L., Van Schaftingen E., Hanson A. D. (2013) Metabolite damage and its repair or pre-emption. Nat. Chem. Biol. 9, 72–80 - PubMed
-
- Van Schaftingen E., Rzem R., Marbaix A., Collard F., Veiga-da-Cunha M., Linster C. L. (2013) Metabolite proofreading, a neglected aspect of intermediary metabolism. J. Inherit. Metab. Dis. 36, 427–434 - PubMed
-
- Rzem R., Veiga-da-Cunha M., Noël G., Goffette S., Nassogne M. C., Tabarki B., Schöller C., Marquardt T., Vikkula M., Van Schaftingen E. (2004) A gene encoding a putative FAD-dependent l-2-hydroxyglutarate dehydrogenase is mutated in l-2-hydroxyglutaric aciduria. Proc. Natl. Acad. Sci. U.S.A. 101, 16849–16854 - PMC - PubMed
-
- Rzem R., Van Schaftingen E., Veiga-da-Cunha M. (2006) The gene mutated in l-2-hydroxyglutaric aciduria encodes l-2-hydroxyglutarate dehydrogenase. Biochimie 88, 113–116 - PubMed
-
- Rzem R., Vincent M. F., Van Schaftingen E., Veiga-da-Cunha M. (2007) l-2-hydroxyglutaric aciduria, a defect of metabolite repair. J. Inherit. Metab. Dis. 30, 681–689 - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
