

Dubowitz syndrome is a complex comprised of multiple, genetically distinct and phenotypically overlapping disorders
- PMID: 24892279
- PMCID: PMC4043752
- DOI: 10.1371/journal.pone.0098686
Dubowitz syndrome is a complex comprised of multiple, genetically distinct and phenotypically overlapping disorders
Abstract
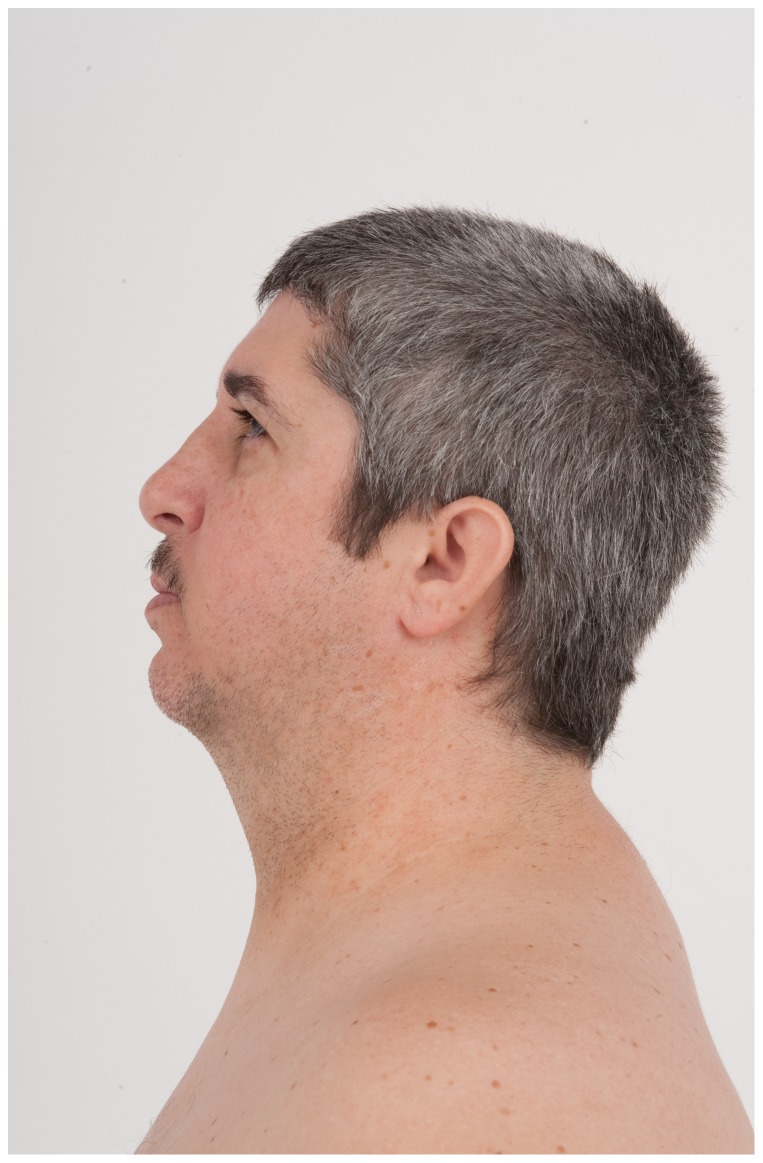
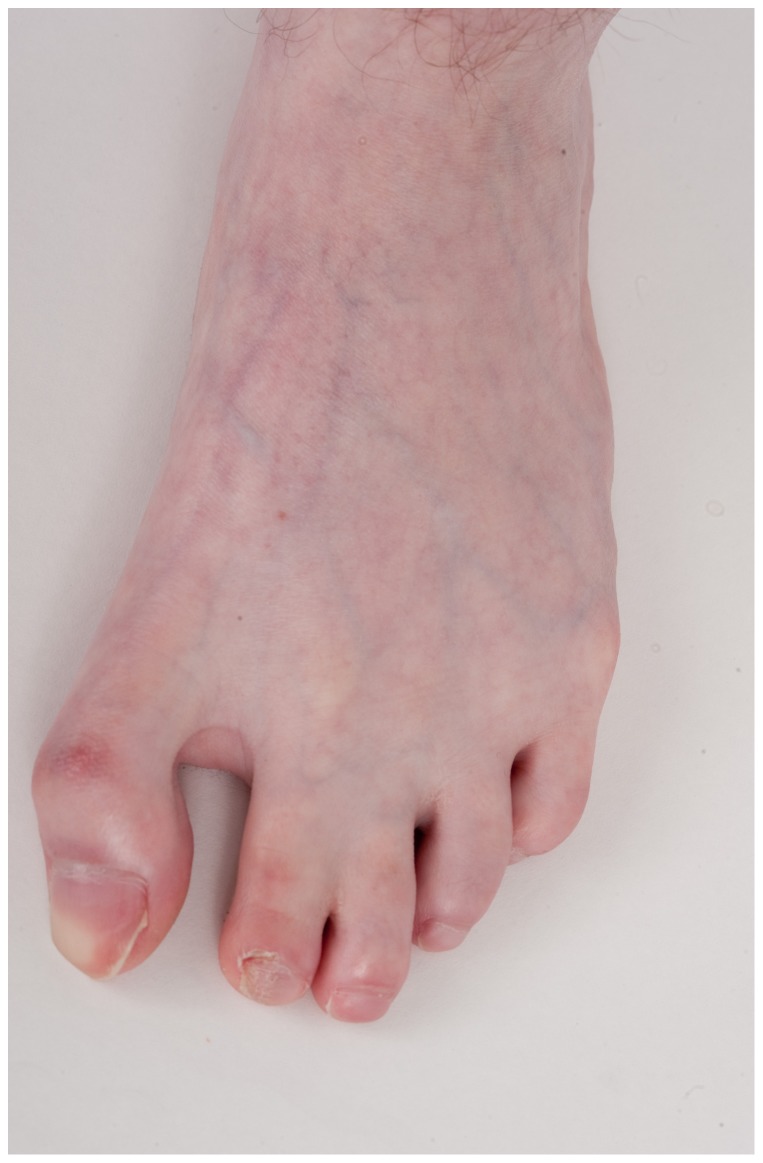
Dubowitz syndrome is a rare disorder characterized by multiple congenital anomalies, cognitive delay, growth failure, an immune defect, and an increased risk of blood dyscrasia and malignancy. There is considerable phenotypic variability, suggesting genetic heterogeneity. We clinically characterized and performed exome sequencing and high-density array SNP genotyping on three individuals with Dubowitz syndrome, including a pair of previously-described siblings (Patients 1 and 2, brother and sister) and an unpublished patient (Patient 3). Given the siblings' history of bone marrow abnormalities, we also evaluated telomere length and performed radiosensitivity assays. In the siblings, exome sequencing identified compound heterozygosity for a known rare nonsense substitution in the nuclear ligase gene LIG4 (rs104894419, NM_002312.3:c.2440C>T) that predicts p.Arg814X (MAF:0.0002) and an NM_002312.3:c.613delT variant that predicts a p.Ser205Leufs*29 frameshift. The frameshift mutation has not been reported in 1000 Genomes, ESP, or ClinSeq. These LIG4 mutations were previously reported in the sibling sister; her brother had not been previously tested. Western blotting showed an absence of a ligase IV band in both siblings. In the third patient, array SNP genotyping revealed a de novo ∼ 3.89 Mb interstitial deletion at chromosome 17q24.2 (chr 17:62,068,463-65,963,102, hg18), which spanned the known Carney complex gene PRKAR1A. In all three patients, a median lymphocyte telomere length of ≤ 1st centile was observed and radiosensitivity assays showed increased sensitivity to ionizing radiation. Our work suggests that, in addition to dyskeratosis congenita, LIG4 and 17q24.2 syndromes also feature shortened telomeres; to confirm this, telomere length testing should be considered in both disorders. Taken together, our work and other reports on Dubowitz syndrome, as currently recognized, suggest that it is not a unitary entity but instead a collection of phenotypically similar disorders. As a clinical entity, Dubowitz syndrome will need continual re-evaluation and re-definition as its constituent phenotypes are determined.
Conflict of interest statement
Figures










References
-
- Opitz JM, Pfeiffer RA, Hermann JP, Kushnick T (1973) Studies of malformation syndromes of man XXIV B: the Dubowitz syndrome. Further observations. Z Kinderheilkd 116: 1–12. - PubMed
-
- Tsukahara M, Opitz JM (1996) Dubowitz syndrome: review of 141 cases including 36 previously unreported patients. Am J Med Genet 63: 277–289. - PubMed
-
- Ahmad A, Amalfitano A, Chen YT, Kishnani PS, Miller C, et al. (1999) Dubowitz syndrome: a defect in the cholesterol biosynthetic pathway? Am J Med Genet 86: 503–504. - PubMed
-
- Hennekam RCM, Krantz ID, Allanson JE (2010) Dubowitz syndrome. Gorlin's Syndromes of the Head and Neck. 5th ed. Oxford: Oxford University Press. pp. 434–436.
Publication types
MeSH terms
Supplementary concepts
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous
