

NOTCH inhibits osteoblast formation in inflammatory arthritis via noncanonical NF-κB
- PMID: 24892805
- PMCID: PMC4071381
- DOI: 10.1172/JCI68901
NOTCH inhibits osteoblast formation in inflammatory arthritis via noncanonical NF-κB
Abstract
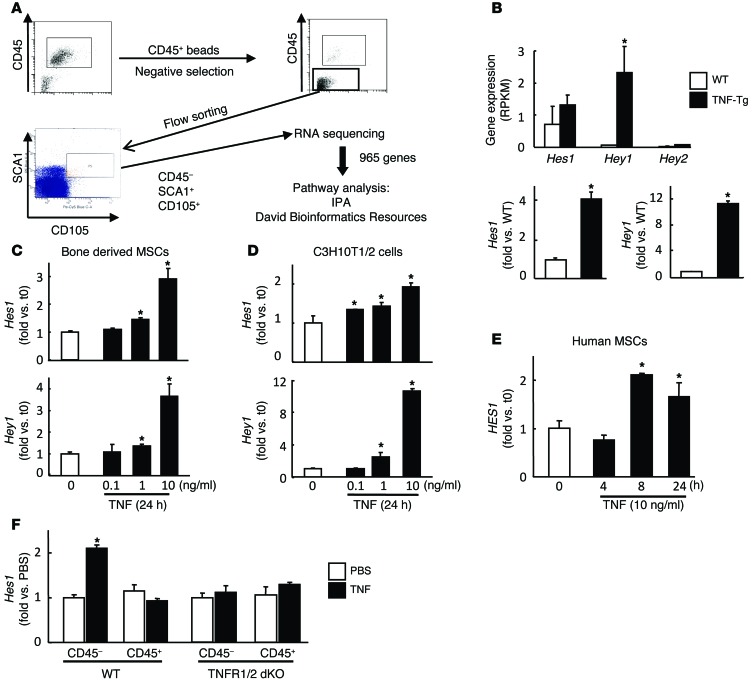
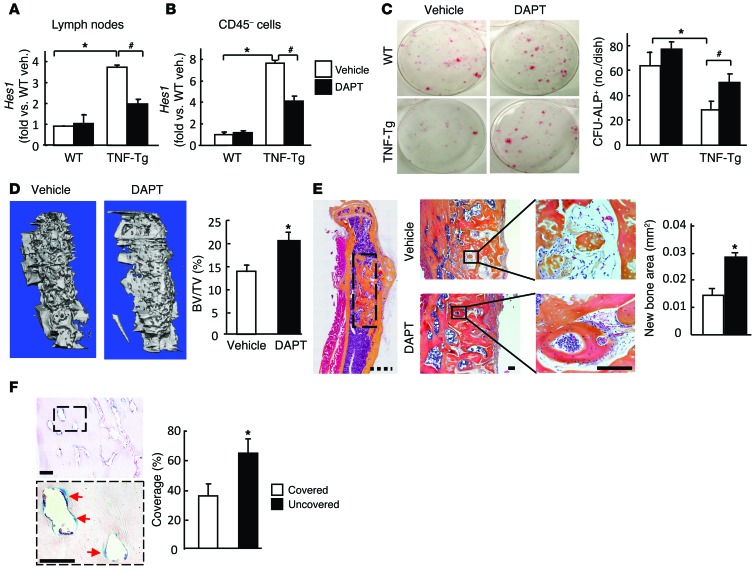
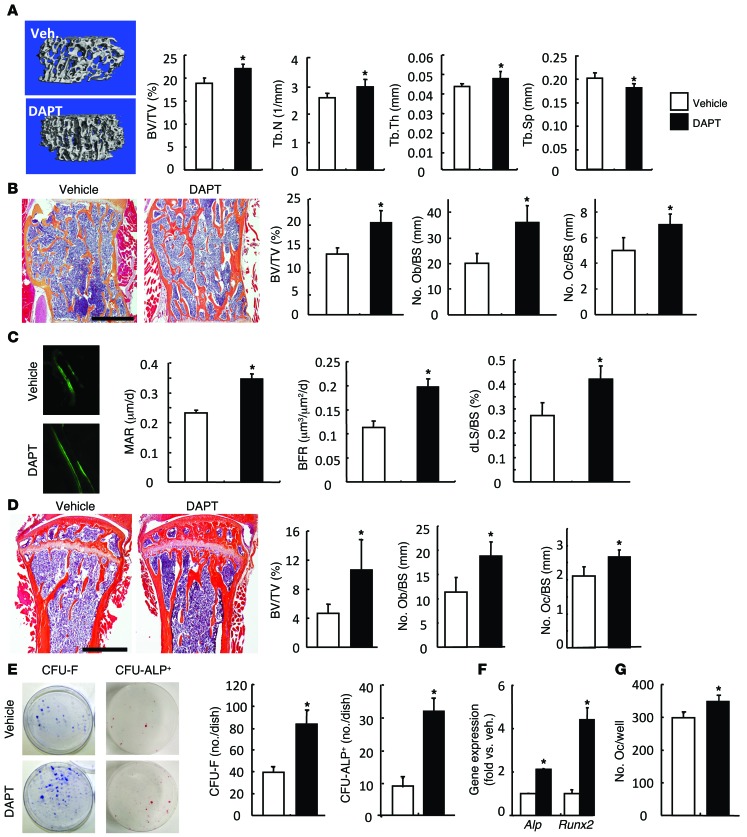
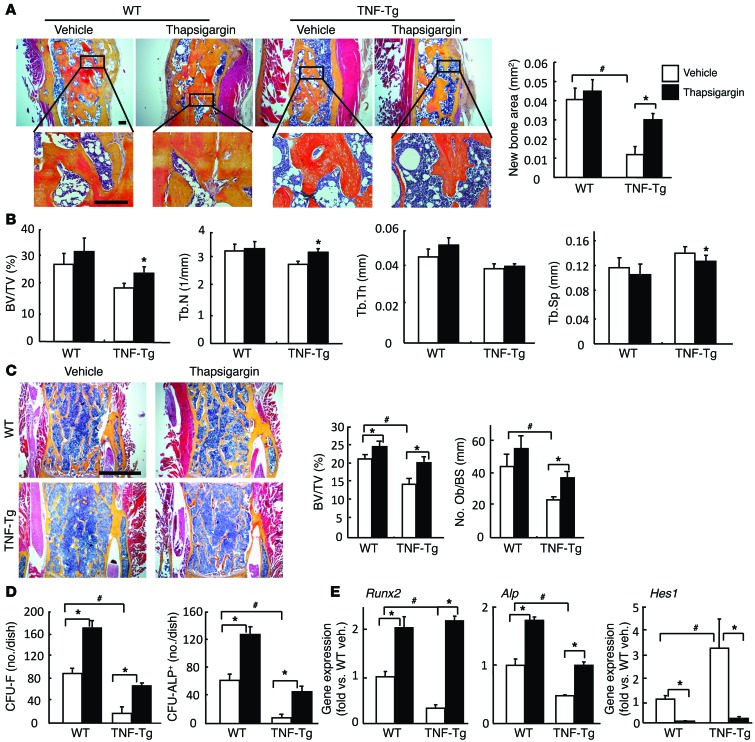
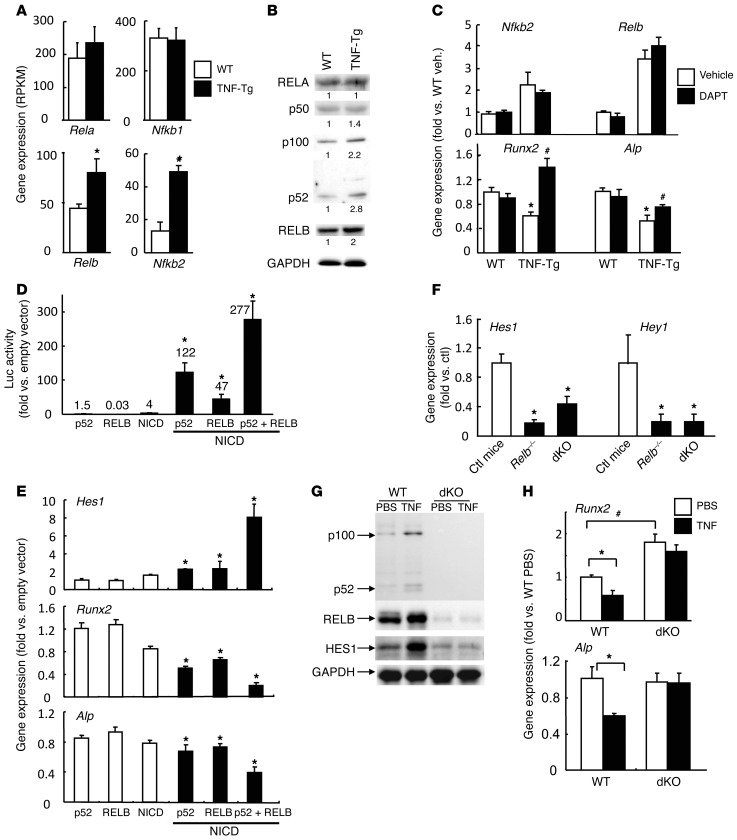
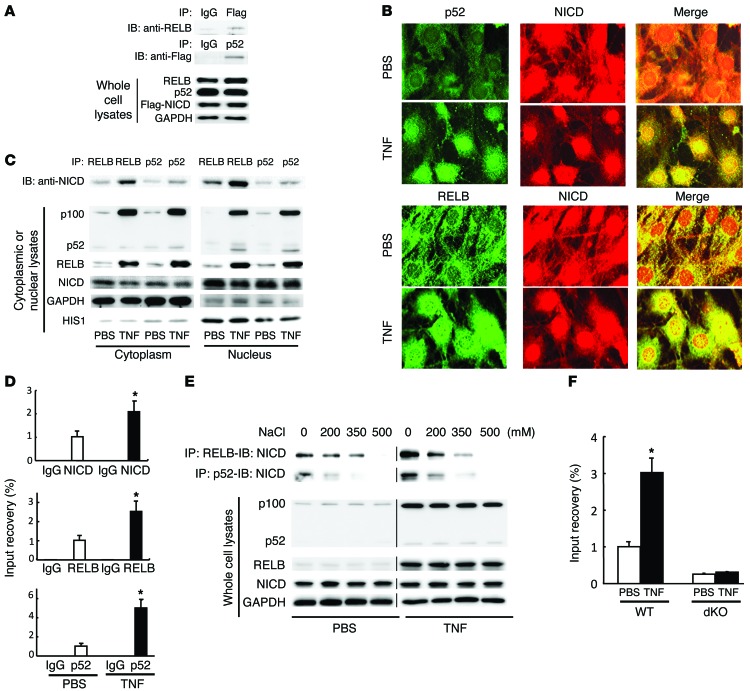
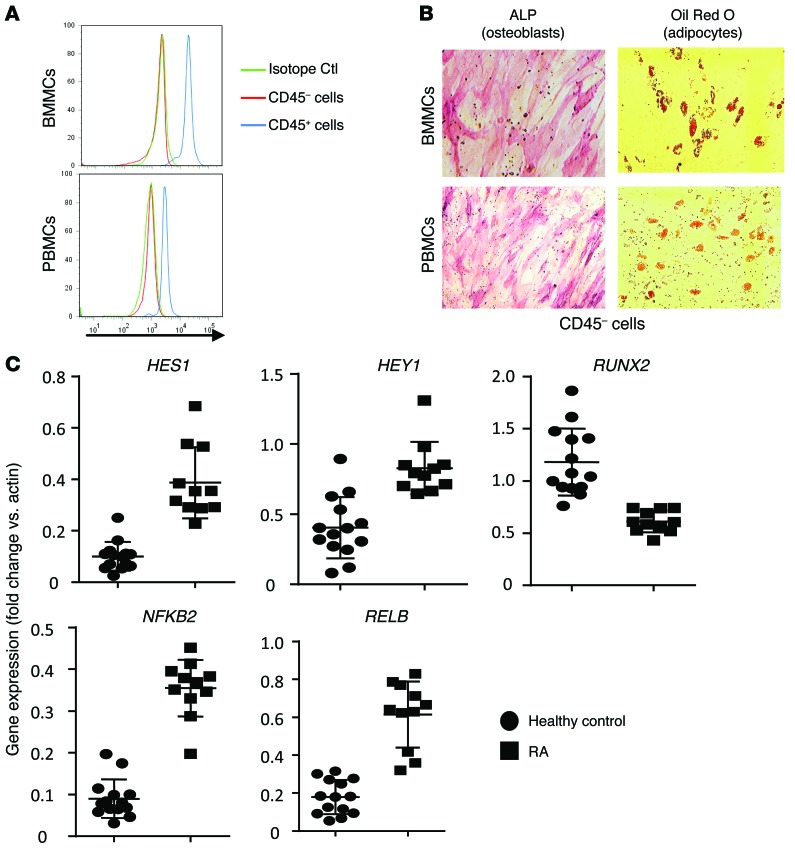
NOTCH-dependent signaling pathways are critical for normal bone remodeling; however, it is unclear if dysfunctional NOTCH activation contributes to inflammation-mediated bone loss, as observed in rheumatoid arthritis (RA) patients. We performed RNA sequencing and pathway analyses in mesenchymal stem cells (MSCs) isolated from transgenic TNF-expressing mice, a model of RA, to identify pathways responsible for decreased osteoblast differentiation. 53 pathways were dysregulated in MSCs from RA mice, among which expression of genes encoding NOTCH pathway members and members of the noncanonical NF-κB pathway were markedly elevated. Administration of NOTCH inhibitors to RA mice prevented bone loss and osteoblast inhibition, and CFU-fibroblasts from RA mice treated with NOTCH inhibitors formed more new bone in recipient mice with tibial defects. Overexpression of the noncanonical NF-κB subunit p52 and RELB in a murine pluripotent stem cell line increased NOTCH intracellular domain-dependent (NICD-dependent) activation of an RBPjκ reporter and levels of the transcription factor HES1. TNF promoted p52/RELB binding to NICD, which enhanced binding at the RBPjκ site within the Hes1 promoter. Furthermore, MSC-enriched cells from RA patients exhibited elevated levels of HES1, p52, and RELB. Together, these data indicate that persistent NOTCH activation in MSCs contributes to decreased osteoblast differentiation associated with RA and suggest that NOTCH inhibitors could prevent inflammation-mediated bone loss.
Figures
Similar articles
-
Use of Hes1-GFP reporter mice to assess activity of the Hes1 promoter in bone cells under chronic inflammation.Bone. 2016 Sep;90:80-9. doi: 10.1016/j.bone.2016.06.003. Epub 2016 Jun 4. Bone. 2016. PMID: 27269414 Free PMC article.
-
Immune Differentiation Regulator p100 Tunes NF-κB Responses to TNF.Front Immunol. 2019 May 7;10:997. doi: 10.3389/fimmu.2019.00997. eCollection 2019. Front Immunol. 2019. PMID: 31134075 Free PMC article.
-
MAML1 regulates cell viability via the NF-κB pathway in cervical cancer cell lines.Exp Cell Res. 2011 Aug 1;317(13):1830-40. doi: 10.1016/j.yexcr.2011.05.005. Epub 2011 May 24. Exp Cell Res. 2011. PMID: 21640102
-
The Role of NF-κB in Physiological Bone Development and Inflammatory Bone Diseases: Is NF-κB Inhibition "Killing Two Birds with One Stone"?Cells. 2019 Dec 14;8(12):1636. doi: 10.3390/cells8121636. Cells. 2019. PMID: 31847314 Free PMC article. Review.
-
Orchestrated modulation of rheumatoid arthritis via crosstalking intracellular signaling pathways.Inflammopharmacology. 2021 Aug;29(4):965-974. doi: 10.1007/s10787-021-00800-3. Epub 2021 Mar 19. Inflammopharmacology. 2021. PMID: 33740220 Review.
Cited by
-
Growth hormone treatment of premature ovarian failure in a mouse model via stimulation of the Notch-1 signaling pathway.Exp Ther Med. 2016 Jul;12(1):215-221. doi: 10.3892/etm.2016.3326. Epub 2016 May 10. Exp Ther Med. 2016. PMID: 27347041 Free PMC article.
-
Non-canonical NF-κB signaling in rheumatoid arthritis: Dr Jekyll and Mr Hyde?Arthritis Res Ther. 2015 Jan 28;17(1):15. doi: 10.1186/s13075-015-0527-3. Arthritis Res Ther. 2015. PMID: 25774937 Free PMC article. Review.
-
The Notch Ligand Jagged1 Regulates the Osteoblastic Lineage by Maintaining the Osteoprogenitor Pool.J Bone Miner Res. 2017 Jun;32(6):1320-1331. doi: 10.1002/jbmr.3106. Epub 2017 Mar 9. J Bone Miner Res. 2017. PMID: 28277610 Free PMC article.
-
Production of RANKL by Memory B Cells: A Link Between B Cells and Bone Erosion in Rheumatoid Arthritis.Arthritis Rheumatol. 2016 Apr;68(4):805-16. doi: 10.1002/art.39489. Arthritis Rheumatol. 2016. PMID: 26554541 Free PMC article.
-
Aberrant Activation of TGF-β in Subchondral Bone at the Onset of Rheumatoid Arthritis Joint Destruction.J Bone Miner Res. 2015 Nov;30(11):2033-43. doi: 10.1002/jbmr.2550. Epub 2015 Jun 8. J Bone Miner Res. 2015. PMID: 25967237 Free PMC article.
References
Publication types
MeSH terms
Substances
Grants and funding
- AR63071/AR/NIAMS NIH HHS/United States
- P30AR0613007/AR/NIAMS NIH HHS/United States
- R01 AI077674/AI/NIAID NIH HHS/United States
- U19AI56390/AI/NIAID NIH HHS/United States
- P01 AI078907/AI/NIAID NIH HHS/United States
- AR43510/AR/NIAMS NIH HHS/United States
- R01 AR043510/AR/NIAMS NIH HHS/United States
- AI077674/AI/NIAID NIH HHS/United States
- P01AI078907/AI/NIAID NIH HHS/United States
- R01 AR063071/AR/NIAMS NIH HHS/United States
- R01 AR063650/AR/NIAMS NIH HHS/United States
- R01 AR048697/AR/NIAMS NIH HHS/United States
- AR48697/AR/NIAMS NIH HHS/United States
- U19 AI056390/AI/NIAID NIH HHS/United States
- AR63650/AR/NIAMS NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
